

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Alzheimer's disease (AD) is the major cause of dementia and the most common form of human amyloidosis. AD affects an astoundingly large number of people and is common with advancing age. The brains of Alzheimer's patients are typically riddled with insoluble “plaques” which consist of amyloid. The major constituent of brain and cerebrovascular amyloid is amyloidα peptide (Aα). A number of genetic, cell biology, biochemical and animal studies support the concept that Aα is central to AD. Here, we discuss regulation of brain Aα and propose that amyloidosis in AD is a “storage” disease caused by inefficient transport of this peptide out of the central nervous system. Thus, we suggest that sporadic AD is at least, in part, a clearance disorder due to defects in transport of Aα that is produced at normal levels throughout the lifetime. The importance of transport-based clearance strategies in conjunction with other Aα-lowering therapies (e.g., immunization/vaccination, Aα-sequestering agents) in preventing the development of cerebral α-amyloidosis and/or clearing toxic clumps from AD brains is discussed.
Introduction
Alzheimer's disease (AD) is the most common form of human amyloidosis and the major cause of dementia. Alzheimer's is common with advancing age. The brains of Alzheimer's patients are typically riddled with insoluble “plaques” which consists of amyloid—small protein fibers that form a hard mass. Neuropathologically, AD is characterized by:
- parenchymal amyloid deposits called neuritic plaques;
- intraneuronal deposits of neurofibrillary tangles;
- cerebral amyloid angiopathy and
- synaptic loss.
The major constituent of the neuritic plaques and congophilic angiopathy is amyloidα peptide (Aα).1 Aα is 4.3 kD peptide that is produced by proteolytic cleavage from a large type 1 transmembrane protein, the amyloidα precursor protein (APP).1
Genetic Risk Factors for AD
Mutations in three genes encoding amyloid precursor protein (APP) on chromosome 21,2,3 presenilin 1 on chromosome 14,4,5 and presenilin 2 on chromosome 16,7,8 have been linked to the rare, early onset autosomal form of AD (onset <60 years). These mutations all affect APP metabolism such that more Aα is produced. In contrast, sporadic AD that represents most AD cases (>98%) has age onset typically above 65 years and exhibits no clear pattern of inheritance (late-onset AD). The ϵ4 allele of the apolipoprotein E (ApoE) gene on chromosome 19 appears to be a risk factor for late-onset AD.9,10 Recently, a susceptibility locus for late-onset AD has been identified on chromosome 1012,13 that acts to increase Aα levels in plasma in the first-degree relatives of patients with typical late-onset AD.14
Amyloid α Peptide
Whether Aα causes or contributes to Alzheimer's dementia is still controversial. A number of genetic, cell biology, biochemical and animal studies support the concept that Aα plays a central role in the development of AD pathology.1,15 Elevated cerebral levels of Aα may occur during normal aging, but the accumulation is significantly accelerated in AD. It is still unresolved how this peptide accumulates in the central nervous system (CNS) and then initiates cytopathology and how much inflammation can contribute to neuronal death. Until recently, significant efforts have been focused on the mechanisms responsible for Aα production, including the roles of the proteolytic enzymes α- and γ-secretases which generate Aα from its APP precursor protein.16,17 Aα is produced by almost all tissues and cells in the body and circulates in biological fluids such as plasma, cerebrospinal fluid (CSF) and brain interstitial fluid (ISF).1 It has been suggested that generation of Aα in the CNS may take place in the neuronal axonal membrane compartment by proteolytic processing of APP after APP-mediated axonal transport of α-secretase and presenilin 1.18
Although increases in Aα production can explain a small percentage of early-onset cases of familial AD bearing inherited mutations in APP or presenilins 1 or 2 genes,19 similar increases in production have not been found in sporadic AD in spite of elevated levels of Aα. It is believed that minimizing physiological production of Aα in sporadic AD by blocking α- and γ-secretases may still reduce accumulation of Aα in the brain. The proof whether Aα causes Alzheimer's dementia or not should ultimately come from clinical trials with Aα-lowering agents. These trials have recently begun including the vaccination of humans with Aα and treatments with γ-secretase inhibitors. However, the results of ongoing trials at present are inconclusive. The vaccination therapy with Aα has been halted due to serious side effects, and there is still uncertainty over the feasibility of long-term therapy with secretase inhibitors16,17 because of their potential effects on other cellular pathways. However, less toxic vaccines and passive immunization are currently under development as well as more selective and specific secretase inhibitors.
A Clearance Mechanism
The realization that increased brain production of Aα is not involved in sporadic AD or late-onset AD led us to propose that amyloidosis in AD can be a “storage” disease caused by inefficient elimination of the peptide from the CNS due to defects in its transport out of the CNS possibly caused by conformational changes in the molecule due to increased concentrations, acidic pH (e.g., inflammation), post-translational modifications (e.g., oxidation, glycation), binding to chaperone molecules and/or downregulation of the blood-brain barrier (BBB) transporters. The mechanisms of Aα clearance have received relatively little attention until recently. The two plausible hypotheses for Aα clearance from the brain are metabolism20,21 and transport out of the CNS,22 as we proposed earlier. Here, we will focus on mechanisms for Aα accumulation caused by inefficient transport of the peptide out of the CNS shortly after its physiological production.
Aα Transport Out of the CNS
The peptide structure of Aα implies that it cannot be eliminated rapidly from the CNS into circulation unless there is a carrier-mediated and/or receptor-mediated transport system(s) for the peptide in brain microvascular endothelium, a site of the BBB in vivo. Although peptides can be cleared to some extent by passive diffusion or transport via a non-specific bulk flow of brain ISF and CSF, this route seems to be responsible for about 10% of Aα clearance from brain tissue under physiological conditions.23 The BBB has unique properties, as it does not normally allow free exchanges of polar solutes such as Aα between brain and blood owing to the presence of tight junctions between brain endothelial cells that form a continuous cellular monolayer.
As shown in Figure 10.1, recent studies have revealed a new role for the low density lipoprotein (LDL), receptor-related protein 1 (LRP1), a member of the LDL receptor family which is central to transport and metabolism of cholesterol and ApoE-containing lipoproteins, as a clearance receptor for Aα at the BBB mediating transport of the peptide from brain into the blood.23 Based on experimentally determined transport kinetic efflux constants, the LRP1-mediated transcytosis of Aα across the BBB is a high-affinity and rate-limiting step for Aα clearance from the brain. The LRP1-mediated Aα transport is initiated at the abluminal (brain) site of the endothelium and is therefore directly responsible for eliminating Aα from brain ISF. The rate of transport is modulated by the LRP1 ligands, ApoE and α2-macroglobulin, that have been identified either as a definite risk factor (i.e., ApoE4)9,11 and/or a possible risk factor for AD,24 respectively. Reduced expression of brain endothelial LRP1 was observed during normal aging in rodents, non-human primates, and AD patients associated with impaired Aα clearance and cerebrovascular accumulation of the peptide.23,25
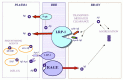
Recent studies have confirmed the role of brain efflux and rapid transport exchanges of Aα between blood and brain across the BBB in non-human primate models of brain parenchymal and cerebrovascular amyloidosis and transgenic models of AD.25 28 It is of note that we were first to suggest in 1993 that the BBB regulates brain Aα via specific receptors and/or transporters;29 the molecular nature of the transport systems and vascular therapeutic targets were not known at that time. It took a decade to characterize at the molecular level different Aα receptors and transporters at the BBB, define their functions and develop molecular reagents and approaches to confirm the role of transport in pathogenesis of amyloidosis in animal models of AD. After our initial report, a series of papers from different groups have verified the validity of the BBB transport hypothesis for Aα.20,23,25-48
Most recent studies indicate that efflux of Aα from brain produces rapid increases in plasma Aα that correlate with the amyloid burden in brain.25,27 It has been suggested that Aα efflux measurements may be useful for quantifying brain amyloid burden in patients at risk for or those who have been diagnosed with AD.27 It has been also demonstrated that development of plaques in non-human primate models of amyloidosis AD type and transgenic animal models of AD shift a transport equilibrium for Aα between the CNS and plasma due to binding of soluble Aα from brain and plasma onto amyloid deposits in the CNS and around blood vessels.25-28,32 Recent studies in squirrel monkeys, a non-human primate model of cerebral amyloid angiopathy, have demonstrated by single photon emission computed tomography a rapid elimination of Aα from the brain into plasma, suggesting significant clearance of the peptide across the BBB.25 Studies in primates also indicated an age-related decline in the BBB capacity to eliminate Aα that correlates with increases in Aα40/42 cerebrovascular immunoreactivity and amyloid deposition.25,26
It has been reported that the P-glycoprotein, a member of the ATP-binding cassette (ABC) superfamily of transporters that remove a variety of lipophilic and amphipatic molecules from cells, may eliminate Aα from brain endothelium via efflux across the apical (blood) facing site of the BBB.47 Thus, the P-glycoprotein cannot directly eliminate Aα from brain ISF but may participate in its efflux from brain endothelium into the circulation. The affinity of P-glycoprotein for the peptide is about 500-fold lower compared to LRP1.
Plasma Aα Transport
The autosomal dominant mutations that cause early-onset familial AD all increase Aα (the 42-amino acids isoform) in plasma and brain.49-53 Recently, it has been shown that a novel late-onset AD locus on chromosome 10 acts to increase plasma Aα.14 At the BBB, LRP234 and the receptor for advanced glycation end products (RAGE)43,54 may transport circulating Aα into the brain in a complex with apolipoprotein J (ApoJ) or as a free peptide, respectively, which could influence Aα accumulation. Under physiological conditions LRP2 is saturated by apoJ, which may preclude the entrance of the peptide into the CNS.34,40 On the other hand, RAGE, a multiligand receptor in the immunoglobulin superfamily, binds soluble Aα in the nanomolar range and can mediate relatively rapid transport of unbound Aα into the brain which may result in significant increases in cerebrovascular Aα if RAGE-mediated influx is not counterbalanced by the efflux from the CNS.43,54
In AD, RAGE is upregulated in the vascular system,55 assuming the role of a “pathogenic” receptor which amplifies Aα accumulation and mediates vascularly induced neuronal stress.54-56 Thus, therapeutic strategies to prevent RAGE-mediated Aα vascular interactions by either blocking vascular RAGE, or by sequestering circulating peptide with a soluble form of the RAGE receptor, sRAGE, may result in reduced amyloid load, as shown in animal models of systemic amyloidosis and AD.57,58 Similarly, it has been recently reported that removing serum amyloid P component (SAP) from human amyloid deposits in the tissue by drugs that are competitive inhibitors of SAP may provide rapid clearance of SAP, thus producing marked depletion of human circulating SAP, which in turn can provide a new therapeutic approach to both systemic amyloidosis and AD.59
Although a key element of Aα accumulation is decreased clearance of brain amyloid due potentially to decreased exit of Aα from the brain, there also appears to be a role for intravascular peptide. We propose that there is a complex, but dynamic, equilibrium at least at an early stage in cerebral amyloid accumulation between pathogenic Aα in the brain and that present in the blood. Trapping Aα in the intravascular space, especially in the form of complexes with sRAGE, SAP or the antibody (see below), appears to promote its clearance from the CNS.
Immunization Strategies and Transport
Recent studies suggested that the mechanism of action of various immunization or “vaccination” approaches to reduce the amyloid burden in AD transgenic mice,60,61 and possibly in humans, involves sequestration of plasma-derived Aα by an anti-Aα IgG antibody.46 According to the proposed transport model in Figure 10.1, blockage of Aα transport into the CNS will not per se increase transport from brain to plasma unless the efflux, i.e., transport out of the CNS, is upregulated. In AD brains, however, the LRP1 at the BBB which mediates Aα transport from brain to blood, is downregulated.23 If the anti-Aα IgG simply mops up the Aα in plasma the influx will fall and with sustained efflux, the cerebral load of Aα would decrease as long as the deposited Aα will resolubilize with time. This implies that the antibody must promote either resolubilization of aggregated Aα in the brain and/or efflux of a rapidly mobilized soluble pool of Aα, that should result in Aα clearing as long the efflux transport systems are intact.
Nonspecific Transport
In addition to the transport systems that rapidly eliminate Aα across the BBB, a nonspecific bulk flow of ISF can also carry Aα and/or its metabolites passively from the ISF into the CSF across the permeable ependyma of brain ventricles, and from the CSF back to blood across the arachnoid granulations, choroid plexus and via drainage into deep cervical lymph. According to recent measurements, the ISFCSF bulk flow has a minor role in physiological clearance of Aα from the CNS.23 However, this route may still be important in removing degraded peptide fragments that are normally not recognized by the Aα putative transporters,36 and/or under pathological conditions when the excess at Aα cannot be cleared across the BBB.
Transport-Based Strategies
An independent validation of the transport-clearance hypothesis has been recently obtained with the Dutch mutant peptide, an isoform of Aα associated with the early-onset familial form of amyloidosis AD type.48 Substitution at codon 22 resulted in reduced Aα clearance from the CNS and CSF due to impaired ability of the mutant Aα peptide to be recognized by the transport systems at the BBB, subsequently leading to its accumulation in the brain.
Only time will confirm whether sporadic AD represents, at least in part, a clearance disorder due to defects in transport of Aα that is produced normally throughout the life, as suggested by numerous experimental studies. Nevertheless, developing new treatments to lower Aα by promoting its transport from the CNS should reduce the cellular stress, spontaneous Aα aggregation, amyloid formation and toxic effects. The body of evidence suggests that transport-based clearance strategies may have potentially important implications for control of cerebral αamyloidosis in AD, and in conjunction with other Aα-lowering therapies including immunization/vaccination, resolubilization of amyloid, blockade of plasma Aα transport and blockade of Aα production, in eliminating amyloid and plaques from the brain.
Acknowledgement
Our research in AD has been supported by PHS grants AG16223 and NS34467.
References
- 1.
- Ghiso J, Frangione B. Amyloidosis in Alzheimer's disease. Adv Drug Del Rev. 2002;54(12):1539–1551. [PubMed: 12453671]
- 2.
- Levy E, Carman MD, Fernandez-Madrid I. et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. [PubMed: 2111584]
- 3.
- Goate A, Chartier-Harlin MC, Mullan M. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. [PubMed: 1671712]
- 4.
- Schellenberg G. Genetic linkage for a novel familial Alzheimer's disease locus on chromosome 14. Science. 1992;258:868–871. [PubMed: 1411576]
- 5.
- Sherrington R, Rogaev EL, Liang Y. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;373:754–760. [PubMed: 7596406]
- 6.
- Levy-Lahad E, Wasco W, Poorkaj P. et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. [PubMed: 7638622]
- 7.
- Rogaev E, Sherrington R, Rogaeva EA. et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. [PubMed: 7651536]
- 8.
- Li J, Ma J, Potter H. Identification and expression analysis of a potential familial Alzheimer disease gene on chromosome 1 related to AD. Proc Natl Acad Sci USA. 1995;92:12180–12184. [PMC free article: PMC40320] [PubMed: 8618867]
- 9.
- Corder EH, Saunders AM, Strittmatter WJ. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. [PubMed: 8346443]
- 10.
- Strittmatter WJ, Saunders AM, Schmechel D. et al. Apolipoprotein E: High-avidity binding to α-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. [PMC free article: PMC46003] [PubMed: 8446617]
- 11.
- Blacker D, Haines JL, Rodes L. et al. ApoE4 and age at onset of Alzheimer's disease: The NIMH genetics initiative. Neurology. 1997;48:139–147. [PubMed: 9008509]
- 12.
- Myers A, Holmans P, Marshall H. et al. Susceptibility locus for Alzheimer's disease on chromosome 10. Science. 2000;290:2304–2305. [PubMed: 11125144]
- 13.
- Bertram L, Blacker D, Mullin K. et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science. 2000;290:2302–2303. [PubMed: 11125142]
- 14.
- Ertekin-Taner N, Graff-Radford N, Younkin LH. et al. Linkage of plasma Aα42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science. 2000;290:2303–2304. [PubMed: 11125143]
- 15.
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–180. [PubMed: 11683988]
- 16.
- Vassar R, Bennett BD, Babu-Khan S. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. [PubMed: 10531052]
- 17.
- Ray WJ, Yao M, Mumm J. Cell surface presenilin 1 participates in the γ-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–36807. [PubMed: 10593990]
- 18.
- Kamal A, Almenar-Queralt A. et al. Kinesin-mediated axonal transport of a membrane compartment containing α-secretase and presenilin 1 requires APP. Nature. 2001;414:643–645. [PubMed: 11740561]
- 19.
- Hardy J, Duff K, Hardy KG. et al. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. [PubMed: 10196523]
- 20.
- Iwata N, Tsubuki S, Takaki Y. et al. Identification of the major Aα1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. [PubMed: 10655101]
- 21.
- Iwata N, Tsubuki S, Takaki Y. et al. Metabolic regulation of brain Aα by neprilysin. Science. 2001;292:1550–1552. [PubMed: 11375493]
- 22.
- Zlokovic BV, Yamada S, Holtzman D. et al. Clearance of amyloid αpeptide from brain: transport or metabolism? Nat Med. 2000;6:718–719. [PubMed: 10888892]
- 23.
- Shibata M, Yamada S, Kumar SR. et al. Clearance of Alzheimer's amyloidα1-40 peptide from brain by LDL receptor-related protein 1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. [PMC free article: PMC387254] [PubMed: 11120756]
- 24.
- Blacker D, Wilcox MA, Laird NM. et al. α2-Macroglobulin is genetically associated with Alzheimer's disease. Nat Genet. 1998;19:357–360. [PubMed: 9697696]
- 25.
- Bading JR, Yamada S, Mackic JB. et al. Brain clearance of Alzheimer's amyloidα40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy J Drug Target 2002. 10 4359–368. [PubMed: 12164385]
- 26.
- Mackic JB, Ghiso J, Frangione B. et al. Differential cerebrovascular sequestration and enhanced blood-brain barrier permeability to circulating Alzheimer's amyloidα peptide in aged Rhesus vs. aged Squirrel monkey. Vasc Pharmacol. 2002;18:3331–3.
- 27.
- De Mattos RB, Bales KR, Cummins DJ. et al. Brain to plasma amyloidα efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. [PubMed: 11910111]
- 28.
- De Mattos RB, Bales KR, Parsadanian M. et al. Plaque-associated disruption of CSF and plasma amyloidα (Aα) equilibrium in a mouse model of Alzheimer 's disease. J Neurochem. 2002;81:229–236J. [PubMed: 12064470]
- 29.
- Zlokovic BV, Ghiso J, Mackic JB. et al. Blood-brain barrier transport of circulating Alzheimer's amyloidα Biochem Biophys Res Commun. 1993;197:1034–40. [PubMed: 8280117]
- 30.
- Maness LM, Banks WA, Podlisny MB. et al. Passage of human amyloidα protein 140 across the murine blood-brain barrier. Life Sci. 1994;55:1643–50. [PubMed: 7968239]
- 31.
- Zlokovic BV, Martel CL, Mackic JB. et al. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloidα Biochem Biophys Res Comm. 1994;205:1431–1437. [PubMed: 7802679]
- 32.
- Ghilardi JR, Catton M, Stimson ER. et al. Intraarterial infusion of [125I]Aα1-40 labels amyloid deposits in the aged primate brain in vivo. Neuroreport. 1996;7:2607–11. [PubMed: 8981432]
- 33.
- Ghersi-Egea JF, Gorevic PD, Ghiso J. et al. Fate of cerebrospinal fluid-borne amyloid α-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880–83. [PubMed: 8764620]
- 34.
- Zlokovic BV, Martel CL, Matsubara E. et al. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer's disease amyloidα at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. 1996;93:4229–4234. [PMC free article: PMC39517] [PubMed: 8633046]
- 35.
- Zlokovic BV. Cerebrovascular transport of Alzheimer's amyloidα and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996;59:1483–1497. [PubMed: 8890929]
- 36.
- Banks WA, Kastin AJ. Passage of peptides across the blood-brain barrier: pathophysiological perspectives. Life Sci. 1996;59:1923–1943. [PubMed: 8950292]
- 37.
- Martel CL, Mackic JB, McComb JG. et al. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer's amyloidα in guinea pigs. Neurosci Lett. 1996;206:157–160. [PubMed: 8710175]
- 38.
- Martel CL, Mackic JB, Matsubara E. et al. Isoformspecific effects of apolipoproteins E2, E3, E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid α J Neurochem. 1997;69:1995–2004. [PubMed: 9349544]
- 39.
- Poduslo JF, Curran GL, Haggard JJ. et al. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid αprotein 140 at the blood-brain barrier. Neurobiol Dis. 1997;4:27–34. [PubMed: 9258909]
- 40.
- Shayo M, McLay RN, Kastin AJ. et al. The putative blood-brain barrier transporter for the αamyloid binding protein apolipoprotein J is saturated at physiological concentrations. Life Sci. 1997;60:PL115–PL118. [PubMed: 9042383]
- 41.
- Zlokovic BV. Can blood-brain barrier play a role in the development of cerebral amyloidosis and Alzheimer's disease pathology? Neurobiol Dis. 1997;4:23–26. [PubMed: 9258908]
- 42.
- Mackic JB, Weiss MH, Miao W. et al. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid αpeptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem. 1998;70:210–5. [PubMed: 9422364]
- 43.
- Mackic JB, Stins M, McComb JG. et al. Human blood-brain barrier receptors for Alzheimer's amyloidβ J Clin Invest. 1998;102:734–743. [PMC free article: PMC508936] [PubMed: 9710442]
- 44.
- Wegenack TM, Curran GL, Poduslo JF. Targeting Alzheimer amyloid plaques in vivo. Nat Biotechnol. 2000;18:868–872. [PubMed: 10932157]
- 45.
- Poduslo JF, Curran GL. Amyloidα peptide as a vaccine for Alzheimer's disease involves receptor-mediated transport at the blood-brain barrier. Neuroreport. 2001;12:3197–3200. [PubMed: 11711855]
- 46.
- De Mattos RB, Bales KR, Cummins DJ. et al. Peripheral anti-Aα antibody alters CNS and plasma Aα clearance and decreases brain Aα burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. [PMC free article: PMC37524] [PubMed: 11438712]
- 47.
- Lam FC, Liu R, Lu P. et al. αAmyloid efflux mediated by P-glycoprotein. J Neurochem. 2001;76:1121–1128. [PubMed: 11181832]
- 48.
- Monro OR, Mackic JB, Yamada S. et al. Substitution at codon 22 reduces clearance of Alzheimer's amyloidα peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging. 2002;23:405–412. [PubMed: 11959403]
- 49.
- Citron M, Oltersdorf T, Haase C. et al. Mutation of the αamyloid precursor protein in familial Alzheimer's disease increases αprotein production. Nature. 1992;360:672–674. [PubMed: 1465129]
- 50.
- Cai XD, Golde TE, Younkin SG. Release of excess amyloidα protein from a mutant amyloidα protein precursor. Science. 1993;259:514–516. [PubMed: 8424174]
- 51.
- Suzuki N, Cheung TT, Cai TT. et al. An increase percentage of long amyloidα protein secreted by familial amyloidα protein precursor (α FF717) mutants. Science. 1994;264:1336–1340. [PubMed: 8191290]
- 52.
- Borchelt DR, Thinakaran G, Eckman CB. et al. Familial Alzheimer's diseaselinked presenilin 1 variants elevate Aα1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. [PubMed: 8938131]
- 53.
- Scheuner D, Eckman C, Jensen M. et al. Secreted amyloidαprotein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. [PubMed: 8705854]
- 54.
- Kumar R, Miao W, Ghiso J. et al. Rage at the blood-brain barrier mediates neurovascular dysfunction caused by amyloidα1-40 peptide. Soc Neurosci Abst. 2000;26:7–41.
- 55.
- Yan SD, Chen X, Fu J. et al. RAGE and amyloidbeta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. [PubMed: 8751438]
- 56.
- Hofman F, Kumar SR, Maness LM. et al. Amyloidα peptide (1-40) reduction in cerebral blood flow is consequent to RAGE-mediated induction of endothelin 1. Soc Neurosci Abst. 2001;27:3–33.
- 57.
- Yan SD, Zhu H, Zhu A. et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. 2000;6:643–651. [PubMed: 10835680]
- 58.
- Yu J, Zhu H, Pettigrew LC. et al. Infusion of soluble RAGE inhibits Aα amyloid deposition in APP transgenic mice. Soc Neurosci Abstr. 2001;27:8–56.
- 59.
- Pepys MB, Herbert J, Hutchinson GA. et al. Targeted pharmacological deletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417: 254–259. [PubMed: 12015594]
- 60.
- Schenk D, Barbour R, Dunn W. et al. Immunization with amyloidα attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. [PubMed: 10408445]
- 61.
- Sigurdsson EM, Scholtzova H, Mehta PD. et al. Immunization with a nontoxic/nonfibrillar amyloid beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. [PMC free article: PMC1850561] [PubMed: 11485902]
- Transport-Clearance Hypothesis for Alzheimer's Disease and Potential Therapeutic...Transport-Clearance Hypothesis for Alzheimer's Disease and Potential Therapeutic Implications - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...