

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Kruger L, Light AR, editors. Translational Pain Research: From Mouse to Man. Boca Raton (FL): CRC Press/Taylor & Francis; 2010.

11.1. INTRODUCTION
Muscle fatigue and pain are among the most common complaints at emergency rooms and clinics across the country. Fatigue and pain are often acute, remitting spontaneously or appearing to be attenuated by a variety of drugs and treatment modalities. In spite of these remissions, popular magazines (e.g., Time) estimate that each year Americans spend over $30 billion on herbal remedies and $50 billion on alternative therapies to treat symptoms that include muscle pain and fatigue. These statistics indicate that even acute muscle pain and fatigue are serious health problems that are not adequately addressed by current medical practice.
Occasionally, muscle pain and fatigue take on a chronic nature, leading to syndromes including chronic fatigue syndrome (CFS) and fibromyalgia syndrome (FMS)—devastating conditions characterized by continuing, debilitating fatigue, which is made worse by even mild exercise in the case of CFS and by chronic widespread pain (CWP) with a particular emphasis in the muscles, which can prevent most or all activities in the case of FMS. Both of these conditions are frequently associated with each other and with a variety of other illnesses, such as temporomandibular disorder (TMD), irritable bowel syndrome (IBS), and multiple chemical sensitivity. These syndromes destroy lives, respond poorly to current treatment strategies, and can lead to exhaustion of the financial resources of afflicted patients. Together, these disorders affect 7 to 20 million people in the United States each year, as reported by various authorities (Reeves et al. 2007). Clearly, patients with these syndromes deserve a concerted research effort to understand, treat, and eventually cure these illnesses.
In contrast to cutaneous pain, which has been thoroughly studied and is comparatively well understood, the molecular mechanisms for muscle pain are still unknown. Even more enigmatic is the symptom of debilitating fatigue. Mosso, in his compendious volume on the subject a century ago, remarked that all cultures seem to have just one word for fatigue (Mosso 1904). Yet fatigue describes many conditions, including failure of muscle fibers to shorten normally, deficient motor command signals, feelings of tiredness, heaviness, pressure, and weakness from muscles, and a feeling of mental fatigue that impedes concentration and performance of conceptual tasks.
The subject of most physiological investigations of fatigue has been voluntary muscle contraction. Decreased function causing failure of voluntary muscle contraction can occur at all levels of the neuromuscular system, including the motor cortex, signaling to motoneurons, motoneuron signals to the muscle, excitation-contraction coupling in the muscle, and actin-myosin filament interactions. However, the most common failure is a decrease in the motor command signal from the motor cortex (see recent reports and reviews by Bellinger et al. 2008; Gibson et al. 2003; Noakes et al. 2005; St Clair and Noakes 2004). A recent review suggests that failures in voluntary muscle contraction are most often caused by a central comparator that integrates homeostatic inputs from many physiological systems and shuts down motor commands when energy resources are threatened (Noakes 2007). One of the homeostatic inputs is suggested to “originate from a difference between subconscious representations of baseline physiological homeostatic state and the state of physiological activity induced by physical activity, which creates a second order representation which is perceived by consciousnessproducing structures as the sensation of fatigue” (Gibson et al. 2003, page 174). We suggest that there is a simpler sensation of fatigue that is triggered by inputs from specific receptors that are sensitive to metabolites produced by muscle contraction. We further propose that this elementary sensation is transduced, conducted, and perceived within a unique sensory system with properties analogous to other sensory modalities such as pain. We call it the “sensation of muscle fatigue.”
11.1.1. Fatigue and Pain as Separate Specific Sensory Systems
What defines a specific sensory system? While Müller (1840) and Bell are credited with the “doctrine of specific nerve energies,” the concept of a specific sensory system has since been modified to require the following three basic elements, at a minimum:
- 1.
A qualitatively unique perception consistently associated with a specific form of energy
- 2.
Specific receptors for the energy or stimulus modality
- 3.
Specific transmission systems and brain regions specialized for transmitting and integrating information from the receptors
11.1.2. Does Pain Satisfy These Requirements?
Pain has been shown to satisfy these three requirements and more (Perl 1971, 1998, 2007). (1) Pain is a unique qualitative experience that is normally not confused with any other sensory experience. While there is not a specific form of energy associated with pain, all peripheral stimuli that induce pain, damage tissue, or threaten tissue damage if the stimulus is sustained constitute a consistent effective form of energy. Therefore, the form of energy is a “damaging form of energy.” (2) The specific receptors for the energy are “nociceptors” that are tuned to energy that can damage tissues. (3) A specific transmission system for pain includes the superficial dorsal horn of the spinal cord, the spinothalamic tract and portions of the thalamus, insular cortex, and cingulate gyri of the brain (Brooks et al. 2005; Craig 2003). Understanding the concept of pain as a specific sensory system has enabled the discovery of molecular receptors that allow sensory endings to be sensitive to the stimuli causing damage and pain. This concept has also led to the discovery of modulating substances, such as opioids that can selectively reduce pain by acting at particular regions of the brain and spinal cord.
11.1.3. Does the Sensation of Muscle Fatigue Satisfy the Tenets for a Specific Sensory System?
(1) Although early experiments documented the sensation of “muscle tiredness or pressure” that occurred with non-painful accumulations of muscle contraction-induced metabolites, the uniqueness of this sensation has not been thoroughly elucidated. (2) Specific receptors for the sensation of muscle fatigue have not been identified. (3) While specific spinal cord pathways for sensations of muscle fatigue have some basis (Wilson et al. 2002), more research is necessary. Similarly, some brain areas have been identified as related to the sensation of fatigue (Caseras et al. 2008; Cook et al. 2004, 2007; Williamson et al. 1997), the most common being prefrontal cortex, insular cortex, and the anterior and/or posterior cingulate gyrus. However, much more work is necessary here as well. Thus, for the sensation of muscle fatigue, satisfaction of the three requirements for serving as a specific sensory system is incomplete.
Sensory fatigue shares many similarities with pain and with dyspnea (air hunger). All are protective sensory phenomena. Pain elicits behavior that protects from tissue injury caused by mechanical, chemical, or thermal insults. Dyspnea protects from increases in CO2 and decreases in O2 that would lead to cell death from anoxia. Fatigue is the first line of defense that protects from the over-utilization of energy stores that would lead to rigor and death. All three of these sensory phenomena activate protective reflexes. Nociceptive input activates withdrawal reflexes, and pain motivates conscious actions that remove the injured body part from the stimulus, and it activates sympathetic reflexes that enhance escape. Dyspnea increases respiratory processes, and it activates sympathetic reflexes to preserve vital functions. Sensory fatigue reduces motor commands, and it activates sympathetic reflexes to increase energy stores by increasing blood flow in working muscles and decreasing blood flow in non-working muscles. Each of these sensations evokes a unique cognitive percept with potent aversive connotations, allowing humans to recognize which system is being affected and engaging behavior to alter the conditions that are causing the sensory experience. So why are dyspnea and pain considered by most scientists to have the characteristics of unique sensory systems, while sensory muscle fatigue is rarely considered as such?
Part of the problem has been semantic. Mosso (1904) promoted and popularized confusion between the sensory phenomenon of fatigue and a definition of fatigue as the loss of ability to contract muscles. The sensory experience of fatigue and the loss of muscle contraction were so strongly interrelated in his writings that they could not be separated, even though he clearly recognized and remarked on a special sensory component of fatigue. This historic emphasis on the failure of muscle contraction as the definition of fatigue has persisted in most influential publications to this day (Davis and Bailey 1997), although some have begun to consider fatigue separate from motor insufficiency, albeit as more of an emotional response than a sensory system (Gibson et al. 2003). The semantics of pain went through a similar evolution with a persistent confusion of sensory phenomena with associated consequences in cognition, memory, emotions, autonomic activation, and motor behavior.
Another problem for both pain and sensory muscle fatigue is that special receptors that encode the sensory signals lack a special, macroscopic external apparatus like an eye, ear, nose, or tongue for us to easily observe. For pain, the special nature of nociceptors was painstakingly demonstrated through careful physiological recording experiments, anatomical demonstrations, and unique psychophysical experiments (summarized by Perl 2007). The molecular characteristics that determined the properties of nociceptors were discovered by the use of convenient special agents such as capsaicin that could be used to selectively activate them. The spinal cord, brain stem, and cerebral pathways and integration centers for pain have been mapped out using procedures that uniquely activate cutaneous nociceptors.
A third problem is that the nature of muscle makes it very difficult to expose sensory receptors that are uniquely associated with muscle pain or sensory muscle fatigue. Exposing muscle receptors for direct activation by dissecting the overlying structures almost inevitably disrupts the circulation and activates nociceptors, confounding any attempts to selectively activate receptors for pain or sensory muscle fatigue. In spite of these problems, many attempts have been made to determine the nature of the sense organs involved in muscle pain and the sensory signals involved in the regulation of sympathetic reflexes that are known to be activated by alterations in muscle metabolites that are associated with muscle contraction. Most of the earliest experiments were indirect, using sympathetic reflexes as readouts for the signals from the sensory receptors in the muscles.
11.2. DO SKELETAL MUSCLES HAVE TWO UNIQUE TYPES OF SENSORY RECEPTORS THAT DETECT METABOLITES: ONE TYPE NOCICEPTIVE (CAPABLE OF SIGNALING PAIN), THE OTHER ERGORECEPTIVE (CAPABLE OF DETECTING MUSCLE WORK)?
More than 70 years ago Alam and Smirk (1937) clearly indicated that both a pre-pain “tiredness” and muscle pain could be evoked by metabolites that were produced by muscle contraction, with the former activated by lower concentrations of metabolites than the latter. Metabolites produced by muscle contraction caused blood pressure increases and the sensations of muscle fatigue and muscle pain.
The initial blood pressure increases (see Figure 11.1) were accompanied not by pain, but by a pre-pain phenomenon described as (1) “tiredness or heaviness but not causing appreciable discomfort.” As metabolites continued to increase the subjects experienced (2) “a period where the sensation in the forearm becomes definitely aching in character and is described as pain which, however, is easily tolerated,” and then (3) “a period with severe pain” follows. They demonstrated that the sensations and the blood pressure increases were mediated by afferent nerve fibers innervating the exercising muscle. Thus, the concept of at least two populations of sensory afferents conveying non-painful versus painful information has been extant for more than 70 years. Subsequent experiments confirmed and refined these observations, clearly demonstrating that contraction-produced metabolites detected by sensory afferents in muscle activate a sympathetic exercise pressor reflex (reviewed by Kaufman and Hayes 2002 and by McCord and Kaufman in Chapter 12 in this volume). The primary neural pathway for this reflex has been assumed to be afferent neurons from skeletal muscle that project to sympathetic centers in the spinal cord. These centers project to the brain stem and hypothalamus, with efferent pathways projecting to sympathetic nuclei in the spinal cord, which project to blood vessels in skeletal muscles.

FIGURE 11.1
Top: Exercise without arrest of the circulation. Bottom: Exercise with arrest of the circulation. Exercise increases metabolites in the working muscles, but also activates sympathetic reflexes that enhance blood flow and reduce metabolites. If the circulation (more...)
An interesting development in this area was the discovery that sensory afferents innervating blood vessels often contain calcitonin-gene-related-peptide (CGRP) (Kruger et al. 1989; Silverman and Kruger 1989). Even more interesting is the hypothesis that CGRP could be released by sensory endings in a “neuroeffector” mechanism, meaning that the sensory endings themselves could directly cause effects on peripheral tissues. Given that CGRP is a potent vasodilator, the activation of muscle sensory afferents could release CGRP that directly evokes the vasodilation part of the exercise pressor reflex that occurs in the working muscle (Micevych and Kruger 1992). A second finding also indicates a very direct effect of sensory afferents on sympathetic output (Papka and McNeill 1993). These mechanisms could greatly enhance or independently control the exercise pressor response if the sensory neurons containing CGRP are responsive to metabolites.
Mense and colleagues demonstrated at least two different populations of muscle receptors in experiments based on mechanical stimulation (see Figure 11.2) (Kniffki et al. 1978, 1981; Mense and Meyer 1985). Most of the non-spindle sensory afferents were group III (corresponding to Aδ afferent fibers in cutaneous nerves) or group IV (non-myelinated fibers corresponding to C-afferent fibers in cutaneous nerves). Some of each group of fibers could be classified as “nociceptors” (responding to tissue-damaging stimuli) or non-nociceptors capable of detecting muscle movements, and therefore possible “ergoreceptors” (receptors capable of detecting muscle work). Recent estimates from Mense’s lab suggest that at least 40% of mechanoreceptive group IV afferents could be non-nociceptive (Mense 2009) (see Figure 11.2).

FIGURE 11.2
Experimental setup and results for muscle Group IV afferent recordings. A: Setup. B: Mechanical threshold determination for Group IV unit. C: Pressure thresholds of 26 LTM (grey) and 26 HTM (black) units. D: Distribution of pressure thresholds of LTM (more...)
Identification of responses to muscle-produced metabolites has been more problematic than characterizing responses to mechanical stimulation. Adreani et al. (Adreani et al. 1997; Adreani and Kaufman 1998) reported that about half of a small sample of Group III and IV muscle afferents responded to metabolites produced by muscle contraction that were likely to be non-nociceptive, while the other half responded to metabolites that would probably elicit a painful response. Attempts to identify the metabolites responsible for activating large populations of muscle sensory afferents have been ambiguous. While numerous substances have been found to activate muscle afferents, when these substances are applied individually to sensory endings in concentrations produced by non-painful or even painful muscle contraction, very few of the afferent endings are activated.
Mosso had determined that substances produced by tetanic muscle contraction could lead to fatigue. He cites his work from 1890 showing that blood from a fatigued animal could cause fatigue-like behavior in an unfatigued animal (Mosso 1904). Mosso inferred from available evidence that fatigue was not caused by a lack of oxygen or increased blood CO2, but it might have been caused by a buildup of lactic acid, a lowering of pH, and a temperature increase. While some of Mosso’s conclusions proved incorrect, mostly because of the lack of knowledge of basic neurophysiology (action potentials, membrane potentials, etc., were unknown at this time), his speculation on the substances that might mediate sensory muscle fatigue were clearly prescient.
11.3. MOLECULAR RECEPTORS THAT ARE ACTIVATED BY METABOLITES PRODUCED BY MUSCLE CONTRACTION
Major advances have been made in the last 10 years in identifying metabolites produced by muscle contraction. The most likely ion channels to detect pH changes, acid-sensing ion channels (ASICs), also called amiloride-sensitive ion channels or amiloride-sensitive ion currents, were cloned and sequenced in 1997 (Waldmann et al. 1997a, b). However, initially it appeared that these channels could not play a role in detecting the normal pH values found in humans. They could not gate enough current to activate sensory endings with the small changes in pH observed in conditions known to cause muscle pain in humans, for example, a pH change from 7.4 to 7.0 that can trigger angina in the heart. Immke and McCleskey (2001a) showed that lactate played a role in enhancing the response of ASICs to pH values between pH 7.4 and 7.0. Thus, lactic acid, at concentrations produced during ischemia and extreme active muscle contractions, was more effective in activating muscle sensory afferents than pH reduction alone, as produced by non-ischemic events (e.g., metabolic acidosis).
Later, another critical co-factor was found. ATP in the low concentrations found in muscle interstitium during normal muscle contractions greatly enhanced the sensitivity of ASICs to pH and lactate (Naves and McCleskey 2005; Yagi et al. 2006). Altogether, these combinations enable the ASIC3 receptor to gate sustained current in a range from pH 7.6–6.6 that is capable of activating sensory endings.
Other researchers in McCleskey’s laboratory showed that ASIC receptors were more abundant in sensory nerve fibers that innervated skeletal and cardiac muscle than skin. The sensory nerve fibers that contained ASICs were found on the outside of small arterioles and venules in the fascia surrounding the muscles and separating fascicle bundles (Connor et al. 2005; Molliver et al. 2005). This location is optimal for detecting levels of muscle produced metabolites as they are being removed by the vascular system. Interestingly, the fascia seems to be the structure most heavily innervated by nociceptors, and most likely involved with eliciting muscle pain in human muscle (Gibson et al. 2009; Takahashi et al. 2005).
11.4. THE NEED FOR TRANSLATIONAL BRIDGES TO FIBROMYALGIA AND CHRONIC FATIGUE SYNDROME
Our group recognized that McCleskey’s data could help answer a number of longstanding questions about how muscle sensory afferents detect muscle work and also signal metabolite-induced muscle pain. First, the results showing that combinations of metabolites were more effective in activating DRG neurons than individual metabolites (Immke and McCleskey 2001b, 2003; Naves and McCleskey, 2005; Yagi et al. 2006) suggested to us a way to understand activation of muscle metaboreceptor sensory neurons. By presenting muscle sensory neurons with various combinations (pairs, triplets, etc.) of the metabolites that are produced by muscle contraction, the metabolites necessary and sufficient to activate the sensory neurons in the fashion they are activated by muscle contraction could be determined. Secondly, once an effective combination of metabolites was determined, the actual molecular receptors utilized by muscle sensory neurons to detect this combination could be confirmed by using selective antagonists to the likely molecular receptors (mostly suggested by Immke and McCleskey 2001b, 2003; Naves and McCleskey, 2005; Yagi et al. 2006). In addition, the identity of the molecular receptors could be verified using PCR and real-time, quantitative PCR to determine if sufficient quantities of mRNA for these specific receptors exist in muscle sensory neurons. Using this one could determine the essential molecular difference between muscle sensory neurons that signal sensory fatigue and those that signal muscle pain.
This knowledge became apparent at the same time that we (A.L. and K.L.) became associated with the Pain Research Center at the University of Utah, headed by Dr. C. Richard Chapman (see Chapter 1). A primary research focus of this center has been the pathophysiology and treatment of fibromyalgia syndrome (FMS), chronic widespread pain (CWP), and chronic fatigue syndrome (CFS)—three syndromes that share chronic pain and fatigue as common symptoms. We recognized that a better understanding of sensory muscle fatigue and muscle pain, as well as sympathetic reflexes activated by muscle afferents, might enhance the understanding of FMS and CFS. Further, a better understanding of FMS could provide insight into the basic nature of sensory muscle fatigue and muscle pain. Because it was apparent that multidisciplinary research would be of great benefit, we formed a larger working group to attempt to determine causes, mechanisms, and treatments for sensory fatigue and muscle pain. The interactions of this group instigated the laboratory animal and translational clinical experiments described below.
To determine if combinations of metabolites might solve the problem of how the exercise pressor response is normally triggered, and possibly also how metabolites trigger the sense of muscle fatigue, we employed calcium imaging of dorsal root ganglion neurons, enabling observation of large numbers of randomly selected sensory neurons. To compare neurons innervating all structures with those innervating muscle, we retrogradely labeled dorsal root ganglion (DRG) neurons from hindlimb muscles by injecting them with the tracer DiI 7–10 days before we harvested the cells. We chose to do these experiments in C57 Bl/6 mice because of availability of mutant mice with deleted genes that putatively contribute to detection of metabolites. (These experiments are described in detail in Light et al. 2008.)
We applied metabolites individually to dissociated DRG neurons cultured for 24 hours as illustrated in Figure 11.3. As shown in this figure even pH 6 (very low physiologically) activated very few DRG neurons, although pH 4, which is definitely out of the physiological range, did activate ~26% of the DRG neurons, as noted by others (Leffler et al. 2006). We also found, as had previous studies, that individual metabolites were relatively ineffective, except for ATP, which could excite up to 60% of DRG neurons when applied at 10μM (Connor et al. 2005; Reinohl et al. 2003). Interestingly, in our experiments, if the normal resting level of ATP outside muscle cells was used to pre-adapt the DRG neurons, very few responses to ATP alone were found when ATP was increased in a graded fashion (see Figure 11.4). This implies that ATP receptors (such as P2X5) sensitive to low ATP concentrations on DRG neurons are normally in an adapted state and incapable of generating activating currents. However, receptors that are activated only by higher concentrations of ATP (e.g., P2X4) can be directly activated by metabolite levels produced by painful levels of metabolites (2 μM, or higher).
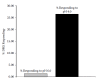
FIGURE 11.3
Percent of all DRG neurons responding to direct application of pH 6.0 of pH 4.0 (from 159 DRG neurons). Acid pH activates few DRG neurons at very low pH (6.0). Non-physiological pH (4.0) is r.
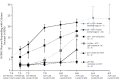
FIGURE 11.4
Percent of DRG neurons responding to various combinations of metabolites. Calcium responses of mouse DRG neurons to various combinations of metabolites at indicated concentrations. Note that neurons are most responsive to the combination of three metabolites (more...)
When two metabolites putatively involved in metaboreception were applied at physiologic levels, only the combination of ATP and protons produced responses that would elicit the sympathetic reflexes observed in humans with increased metabolites (compare Figure 11.4 with Figure 11.1). However, when ATP, protons, and lactate were applied in a graded manner, similar to the way in which they would increase with exercise in a circulation occluded muscle, the responses of DRG neurons, and particularly of muscle-innervating DRG neurons (determined by retrograde labeling), mimicked very closely the sympathetic responses used by others as a read-out of the metaboreceptive responses (Figure 11.4).
Some DRG neurons responded to low levels of metabolites, while others responded best to high levels of metabolites (see Figure 11.5). When we graphed the average amplitude of the calcium responses, the results revealed response profiles of the low-metabolite-responding neurons that were consistent with signaling that elicits the exercise pressor response. These profiles also fit best with perceptions of muscle tiredness that accompany increases in muscle metabolites.

FIGURE 11.5
Responses of DRG neurons to application of indicated levels of metabolites.
The responses of the high metabolite responders, on the other hand, fit best with perceptions of pain that are caused by greater increases in muscle metabolites (see Figure 11.6).
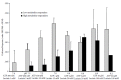
FIGURE 11.6
Calcium response of muscle-labeled DRG neurons responding to changes in metabolites indicated at bottom of graph. (Reprinted with permission from Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J. 2008. Dorsal root ganglion neurons innervating skeletal (more...)
To determine if the molecular receptors mediating calcium responses were those suggested by McCleskey’s group, we determined the metabolite-induced responses of DRG neurons innervating muscle when selective antagonists were applied. These experiments demonstrated that the most likely molecular receptors mediating responses to metabolites are an ASIC receptor (likely ASIC3), a P2X receptor (likely both P2X5, and P2X4), and surprisingly, TRPV1 (see Figure 11.7).

FIGURE 11.7
Percent of DRG neurons responding to metabolites at values indicated at bottom of graphs when ASIC and TRPV1 antagonists were co-applied. (Reprinted with permission from Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J. 2008. Dorsal root ganglion (more...)
The inclusion of TRPV1 (half of the metabolite responsiveness was abolished by TRPV1 antagonists) was surprising, because pH responses at 6.6 are unlikely to be mediated by activation of this receptor. TRPV1 has been shown to be inactive until lower pH levels are reached (>pH 6.0), but we had previously noted that most of the pH-responsive neurons labeled from muscle were responsive to capsaicin, indicating they probably express functional TRPV1 receptors. But what is the functional role of TRPV1 receptors on DRG neurons if not the detection of activation by protons? One possibility is detection of endogenous vanilloids, like anandamide (Cristino et al. 2008; Toth et al. 2005). They also might detect muscle temperature, which like the metabolites is a by-product of muscle contraction. If activated by temperature, the TRPV1 receptors may act like P2X5 receptors in McCleskey’s experiments and may not contribute directly to excitation, but rather enhance the responses of DRG neurons when muscle temperature rises above normal body temperature. Sugiura et al. (2003) demonstrated a similar phenomenon in DRG neurons—that pH 6.0-evoked, sustained currents were enhanced by increasing temperature. Interestingly, we found that low-metabolite-detecting DRG neurons responded to capsaicin just as frequently as high-metabolite-detecting neurons. In addition, the responses to low metabolites were partially blocked by the TRPV1 antagonist LJO 328, implicating TRPV1 receptors in these responses. Thus, temperature could be a factor in activation of muscle “fatigue receptors” as well as nociceptors. This could explain the increased sensory fatigue caused by increased muscle and core temperatures (Drust et al. 2005). Increased blood flow generated by sympathetic reflexes activated by fatigue receptor stimulation would help moderate muscle temperature increases and would decrease the other metabolites generated by muscle contraction.
Are other metabolites and receptors involved in signaling sensory muscle fatigue and pain? McCleskey (personal communication) found that the precursor molecule pyruvate could substitute for lactate but was only ~50% as effective in enhancing metabolite responsiveness. Lactate does not act alone and enhances responsiveness in the most common range of metabolites, but the fact that pyruvate can act as a substitute might explain recent experiments showing that sympathetic reflexes evoked by contracting muscle were blocked in patients with McCardle’s disease (genetic loss of ability to express lactate in muscle while preserving pyruvate) under some conditions, but present under other conditions (see review by Kaufman 2003).
Several other metabolites associated with muscle contraction have been suggested to be important in signaling that elicits the exercise pressor reflex and/or muscle pain. These include ammonia, mono or di-protonated phosphate, CO2, ADP, adenosine, prostaglandins, and potassium. We tested ammonia and ammonium (the species most abundant at low pH values), CO2, and ADP and found them to be ineffective or inhibitory in directly activating muscle-innervating DRG neurons or in enhancing responses to the metabolites shown in the figures above. We assume that the effects of potassium would be non-specific, as increases in extracellular potassium increase the excitability of all neurons, as noted by (Rybicki et al. 1985). We have not yet tested the effects of increased phosphate, adenosine, or prostaglandins, leaving open the possibility that they could contribute to the normal sensations of muscle fatigue and pain and to the exercise pressor response.
Retrospectively, the metabolites we found to synergistically activate a combination of molecular receptors suggests a unique detection system for the rapid use of energy stores, which can signal the body to protection against metabolic and traumatic injury. The combination of protons, lactate, ATP, and temperature increases seems especially relevant to muscle contraction and also applies to brain function. A false signal of sensory muscle fatigue or muscle pain would not occur with metabolic acidosis or respiratory acidosis, as only protons would be increased in these conditions, which would be ineffective in activating muscle afferent neurons unless extreme. On the other hand, traumatic muscle injury, which would cause release of high concentrations of ATP, would activate muscle nociceptors and fatigue receptors, leading to protective behavioral effects (decreased muscle contraction and removal of the injured muscle from the source of trauma).
11.5. ARE THESE MOLECULAR RECEPTORS RESPONSIBLE FOR SENSORY MUSCLE FATIGUE AND MUSCLE PAIN?
As part of a larger investigative group interested in muscle pain and sensations related to muscle fatigue, we recognized that these are greatly increased in people following exhausting exercise. Muscle pain and sensory muscle fatigue ameliorate quickly once exercise is ended, but increase again in untrained muscles 12–24 hours later and can last 48 hours or longer before diminishing. The pain has been studied extensively and is often called delayed-onset muscle soreness (DOMS), and most of us (having experienced it personally) know that a sense of muscle fatigue is also increased during DOMS. The explanation for DOMS has been that exercise of untrained muscle causes micro-tears that result in inflammation; however, this explanation is controversial (Cheung et al. 2003). Whether or not micro trauma is involved, the sensory receptors encoding muscle pain and fatigue must be sensitized in order for these enhanced sensations to be apparent because the metabolite levels are not increased at rest in DOMS. As increases in ASIC3 have been shown in some models of long-term muscle pain (Voilley et al. 2001), we hypothesized that one or more of the molecular receptors involved in metabolite signaling of muscle pain would reveal increased mRNA (possibly leading to increased number of receptors, and therefore, increased signaling) following exercise that produced DOMS.
We exercised mice by running them in hamster balls until they would not run any longer (~2 hours), then sacrificed the mice 16 hours later and collected the DRGs from cervical, thoracic, and lumbar regions of the spinal cord. We hypothesized that increases in mRNA for ASIC3, P2X5, P2X4, and/or TRPV1 would be observed in cervical and lumbar DRG neurons innervating the forelimb and hindlimb muscle groups exercised extensively by running.
As Figure 11.8 shows, mRNA for ASIC3, P2X4, and P2X5 were increased in those lumbar and cervical DRG neurons innervating the exercised limbs. Increases were much smaller in the thoracic region, which contains few neurons innervating the limbs. We interpret this to mean that ASIC3, P2X4, and P2X5 contribute to the enhanced pain and sensory fatigue experienced during DOMS, and possibly during other events that cause long-term increases in muscle pain and sensory muscle fatigue.

FIGURE 11.8
Mouse DRG mRNA increases caused by exhausting exercise, measured 16 hours after a bout of exercise (n = 6).
Several other mediators are also likely to be involved in enhancement of pain and fatigue caused by inflammatory conditions. These include products of the arachidonic acid pathways (Hayes et al. 2006), cytokines (Hoheisel et al. 2005), and neuropeptides such as CGRP (Ambalavanar et al. 2006, 2007; Kruger et al. 1989; Micevych and Kruger 1992). Further work is needed to determine all the contributors to this process.
11.6. TRANSLATION FROM MOUSE TO HUMAN
The next question was, do the molecular receptors we identified in mouse play a role in the human sensory phenomena of muscle pain and fatigue? This presented a problem because sensory muscle fatigue and muscle pain induce the same behavior in mice: They quit running. Thus, discriminating sensory muscle fatigue from muscle pain in the rodent model is difficult. In human patients with CFS, fatigue is the defining symptom, while muscle pain is a common secondary symptom. In patients with FMS or CWP, widespread muscle pain is the defining symptom, and fatigue is the most common secondary symptom. These seemed like appropriate human models to test the idea that ASIC, P2X, and TRPV1 receptors were involved in muscle pain and muscle tiredness. The question, however, was how to access the sensory afferents.
We knew that leukocytes in human subjects show changes and accumulate in the exercised muscle in some cases following exhausting exercise (Kruger et al. 2008). In order to do so, these cells must detect a signal from muscle, and we hypothesized that this signal might be the same metabolites detected by afferent neurons innervating muscle. These leukocytes might utilize the same molecular receptors to detect the metabolites.
Analysis of mRNA from mouse blood indicated that all the molecular receptors we had found to be important for detecting increases in metabolites in muscle sensory neurons were present in leukocytes, and a similar mRNA analysis of human blood also showed that human leukocytes had substantial quantities of these receptor mRNAs. Was the mRNA in leukocytes altered by exercise? This proved to be difficult to determine in mice, because the amount of blood we could obtain was insufficient to measure the mRNA of these genes accurately. On the other hand, sufficient quantities of blood could easily be obtained at multiple times from human subjects. We evaluated mRNA increases in blood from human subjects after exercise and measured the homologous mRNA of ion channels that had been altered in DRG neurons innervating mouse muscle and also mRNA for α2A, β1, and ($β2 adrenergic receptors, catechol-o-methyl transferase (COMT) and several pro-inflammatory and antiinflammatory cytokines and cytokine receptors that might be altered by exercise.
Increases in mRNA obtained from leukocytes from a healthy subject who performed maximal, exhausting strength and aerobic whole-body exercise for 45 minutes (Figure 11.9) were found for the same molecular receptors important for detection of metabolites in mouse DRG neurons that were increased by exhausting exercise in mouse DRGs. Clearly, mRNA increases were strongly related to the timing of DOMS and sensory muscle fatigue that appeared in this subject following exercise.
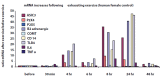
FIGURE 11.9
(see color insert following page 166) mRNA increases in human leukocytes at times indicated after 45 minutes of exhausting exercise. All data are from one female subject. Exercise consisted of aerobic and strength exercises including indoor rock climbing, (more...)
11.7. MOLECULAR RECEPTORS INVOLVED IN CHRONIC FATIGUE SYNDROME
The preceding observations encouraged us to determine if the mRNA of sensory muscle fatigue and pain receptors was increased in CFS patients following exercise that exacerbates their symptoms. A major hallmark of CFS is that even mild exercise greatly increases sensory muscle fatigue as well as muscle pain (Holmes et al. 1988). With guidance from Andrea White, our exercise scientist, we developed a 25-minute, moderate, whole-body exercise protocol in which each subject performed continuously at 70% of his/her age-predicted maximal heart rate. We confirmed that in normal, healthy women and men, this moderate exercise produced minimal muscle pain or sensory muscle fatigue during or immediately after completion, and DOMS did not develop. We compared healthy controls versus patients with CFS and FMS, collecting leukocytes and extracting their mRNA at five intervals: at pre-exercise baseline, and at 30 minutes, 8, 24, and 48 hours after the end of exercise to examine ASIC3, P2X4, P2X5, TRPV1, adrenergic α2A, adrenergic β1β1 and ββ2, COMT, IL6, IL10, TNF-ctα, TLR4, and CD14.
The results (Figure 11.10; Light et al., 2009a) show that following moderate exercise, control subjects revealed no significant increase in mRNA for any of the genes tested. CFS patients (70% with chronic widespread pain meeting diagnostic criteria for FMS) exhibited significant increases in mRNA expression of nearly all of the genes studied, except for IL6 and IL10. The surprise was that in CFS patients, increases in metabolite-detecting receptor and adrenergic receptor gene expression were obvious at 30 minutes after onset of exercise. A second surprise was the magnitude of the effects that greatly exceeded any increases we had seen even with maximum exercise in control subjects. In addition to the mean differences, we found strong correlations between increases in mRNA of ASIC3, P2X4, P2X5, TRPV1, adrenergic α2Aα2A, adrenergic β1, β2, COMT, IL10, TLR4, and CD14 in the CFS patients and their reports of increased mental fatigue during the 2-day time period following exercise. We found similarly strong correlations between ASIC3, TRPV1, adrenergic α2Aα2A and ($β2, and IL10 and the patients’ reports of pain during the 2 days following the exercise challenge.

FIGURE 11.10
(see color insert following page 166) mRNA increases in CFS patients after 25 minutes of moderate exercise (70% of age adjusted maximum heart rate) (middle) but not in control subjects (top). Much more intense exercise (85% of age-adjusted maximum heart (more...)
These data indicate that a common transcription factor is likely altered in CFS, causing strong upregulation of mRNA production in leukocyte genes that detect metabolite increases, and presumably affecting leukocyte production of cytokines. Whether similar action takes place on muscle sensory neurons in CFS patients is unknown, but seems likely, given information suggesting that peripheral signals for pain can be enhanced in at least some of these patients (Staud et al. 2003).
11.8. DYSREGULATED MRNAS MAY ALSO EXPLAIN COMMON CO-MORBIDITIES OF CFS
Many of the genes we found to be dysregulated in CFS patients after exercise may also be implicated in related disorders. In our CFS patients, 70% met criteria for FMS, which is characterized by widespread pain dominated by muscle pain. The muscle nociceptors found in our mouse studies utilized most of the molecular receptors upregulated in CFS patients, including ASIC, P2X4, and TRPV1. Colon sensitivity to inflammation has been shown to be mediated by ASIC receptors and TRPV1 receptors (Christianson et al. 2006; Jones III et al. 2005; Page et al. 2005; Sugiura et al. 2007), suggesting that dysregulation of these receptors could increase the likelihood of irritable bowel syndrome (IBS), another condition often co-morbid with CFS. Multiple chemical sensitivity may be mediated by a TRP channel, possibly TRPV1 or TRPA1 (Pall and Anderson 2004).
Several sensory modalities (but not all) become more sensitive in some cases of CFS. Hyperacusis in CFS may be related to increased ASIC receptors (Geisser et al. 2008; Hildebrand et al. 2004; Peng et al. 2004), as ASIC receptors appear to enhance hearing. Clinical depression may share some of the genes that were upregulated in CFS patients; for example, TRPV1 has recently been implicated in depression (Di et al. 2008), and ASICs may play a role in depression and anxiety (Coryell et al. 2007; Wemmie et al. 2003, 2004). In addition, polymorphisms in COMT may be risk factors for depression, although this issue is not without controversy (Baekken et al. 2008; Funke et al. 2005; Ohara et al. 1998; Tsai et al. 2009). Perhaps genes contributing directly to the sensory experience of fatigue share upstream regulation with the genes that are modified in these co-morbid conditions that share sensory muscle fatigue, hyperalgesia, and chronic pain as central symptoms, and some of the same regulated genes.
A number of other conditions that exhibit unexplained sensory muscle fatigue may also share dysregulation of some of these same sensory, adrenergic, and cytokine genes. We have preliminary evidence that unexplained fatigue in multiple sclerosis, for example, is associated with baseline changes in TRPV1 and adrenergic β2 receptors as well as alterations in several of the ion channel genes and adrenergic receptors following moderate exercise. A similar dysregulation might also explain the greatly enhanced sensory muscle fatigue often observed in heart failure, even when the vascular dynamics appear to be adequate for perfusion (Falk et al. 2006; Khan and Sinoway 2000). If the cardiac sensory receptors, for example, were dysregulated by primary insult to the heart, a profound sensation of fatigue might ensue, unattributable to any particular muscle.
11.9. CAN ACUTE FATIGUE LEAD TO CHRONIC FATIGUE?
A key question about CFS is whether it is similar to chronic pain in the sense that acute pain might constitute a cause of chronic pain (Vierck, Jr., 2006). A clue in this regard would be whether receptors that detect metabolites associated with sensory muscle fatigue appear to be under positive feedback control. Activation of ASICs, P2X, and TRPV1 increases the mRNA of these receptors in dorsal root ganglia of mice following exhausting exercise, and indeed they are upregulated following acute activation of these receptors by metabolites, possibly resulting in more robust activation by future episodes of metabolite production. If the mRNA increases we have observed in normal subjects and particularly in CFS patients are translated into functional receptors, further production of metabolites will produce more receptors, causing an increase in the signal for muscle sensory fatigue if compensating mechanisms are absent (for example, rest, increased sympathetic effects on vascular smooth muscle to eliminate metabolites, and increased capillary capacity—all of which will reduce metabolites). It is also possible that sensory muscle fatigue is like pain in that persistent sensory input may cause central “wind-up” and other plastic changes (such as glial activation; Chacur et al. 2008) that can enhance the afferent signals at many levels in the central nervous system. Thus, sensory experiences for both fatigue and muscle pain could be enhanced over time, and the sympathetic signals caused by sensory muscle fatigue and muscle pain could increase centrally in the spinal cord and brain. There is considerable evidence that signals for fatigue and muscle pain are enhanced in CFS and FMS patients (Caseras et al. 2008; Cook et al. 2007; Geisser et al. 2008; Gracely et al. 2002; Schmaling et al. 2003), although the relative contribution of central components versus peripheral components remains undetermined.
11.10. COMMON CAUSES OF ACUTE FATIGUE MIGHT CAUSE UP-REGULATION OF THE mRNA FOR THE MOLECULAR RECEPTORS SUGGESTED TO MEDIATE SENSORY MUSCLE FATIGUE AND PAIN
At present, we do not know all of the factors that cause upregulation of the molecular receptors responsible for detecting contraction-produced metabolites in muscle sensory neurons. Mamet et al. (2003) provided evidence that nerve growth factor (NGF) can maintain and upregulate ASIC receptors and can mediate inflammatory effects on ASIC receptor upregulation. Thus, any event that increases NGF could increase sensory muscle fatigue and pain.
Cytokine receptors have also been demonstrated on sensory neurons, (e.g., Li et al. 2004). As cytokines have been implicated in the symptoms of a number of viral infections, including Epstein-Barr (Wright-Browne et al. 1998), it is quite likely that viral infections induce muscle fatigue and muscle pain by sensitizing the sensory muscle afferents that signal muscle fatigue and pain. Whether these effects include upregulation of the molecular receptors that encode metabolites is unknown but also seems likely. Thus, acute viral infection could cause myalgia and fatigue through cytokine pathways.
Reductions in mitochondrial efficiency (viral or non-viral causes) could result in decreased aerobic capacity. These decreases could cause increased glycolysis, which would lead to increases in the metabolites that activate sensory muscle afferents. Any such increases could also lead to increased resting and exercise sensations of muscle fatigue and pain. Such increases could also lead to increases in the molecular receptors that encode metabolites, causing further increases in muscle pain and fatigue, and eventually leading to sympathetic dysregulation and possibly CFS and FMS. Clearly, further investigation of the mechanisms that can cause increases in metabolite-detecting receptors is necessary and could clarify a host of conditions in which muscle fatigue and pain are major symptoms.
11.11. MENTAL FATIGUE
Although beyond the scope of this report, similar processes could lead to dysregulation of receptors encoding mental fatigue. Of course, given that the metabolic demands of the brain are different from those of muscle, the specific molecular receptors encoding brain metabolites and the way these receptors regulate cerebral blood flow may differ. However, recent reports suggest there is dysregulation of global cerebral blood flow in CFS patients (Mathew et al. 2008; Yoshiuchi et al. 2006) that could lead to metabolite increases similar to those identified in skeletal muscle of CFS patients.
11.12. SYMPATHETIC DYSREGULATION CONTRIBUTES TO ENHANCED SENSORY FATIGUE
The muscle sensory afferents that may encode sensory muscle fatigue have been linked to sympathetic responses caused by muscle contraction for at least 70 years (Alam and Smirk 1937; see also discussion in Light et al. 2008). Thus, sensory fatigue afferents (and possibly muscle pain afferents) may be the sensory arm(s) of sympathetic reflexes associated with muscle contraction. These reflexes normally serve to increase the blood flow to working muscles partly by diverting blood flow from non-working muscles. This increased blood flow serves to reduce metabolites, terminating the signals underlying sensory muscle fatigue. The sympathetic output signal is mediated by adrenergic β2 receptors on vascular smooth muscle in the working muscle and by adrenergic α receptors in non-working muscles, and by adrenergic β1 receptors in heart muscle.
If the sympathetic signal is not correctly regulated, increased metabolites will persist, causing a constant signal from “fatigue” sensory afferents. Potentially, a constant signal of fatigue from sensory afferents could lead to sympathetic dysregulation by sending a constant sympathetic signal to adrenergic β receptors on vascular smooth muscle in skeletal muscles leading to down-regulation of these adrenergic P receptors. Such downregulation could lead to enhanced metabolites in working muscles, causing an enhanced signal for sensory muscle fatigue from virtually all muscles in the body. This enhanced signal could lead to further dysregulation of sympathetic reflexes. Similarly, if the sympathetic signal is not correctly terminated, for example if COMT is dysregulated either by environmental or genetic causes (Goertzel et al. 2006) leading to increased norepinephrine and epinephrine action on vascular adrenergic βreceptors, down regulation can occur, and the metabolites may not be regulated appropriately. Similarly, alterations in the effectiveness of adrenergic receptors (as is the case in some polymorphisms) could lead to a mismatch between the sensory signal sent to the sympathetic nervous system, and the amount of vasodilation and/or vasoconstriction that results leading to dysregulation of muscle metabolites (Diatchenko et al. 2006). This dysregulation of muscle metabolites could lead to enhanced sensory muscle fatigue both at rest and following exercise.
11.13. ADRENERGIC RECEPTORS ON SENSORY NEURONS?
A dilemma concerning the hypothesized dysregulation of sympathetic reflexes in CFS and FMS patients derives from the view that dysregulation results from an overactive sympathetic nervous system that “burns out” because of desensitization or downregulation of peripheral receptors in vascular smooth muscle resulting from constant bombardment with norepinephrine. In a double-blind, placebo-controlled crossover experiment designed to observe this dysregulation, we (Light et al. 2009b) intravenously administered a low dose of the non-selective β adrenergic antagonist propranolol or placebo to patients with FMS or temporomandibular disorder (TMD). Surprisingly, the sympathetic reflexes (assessed by plasma catecholamine and cardiovascular responses to various challenges) that were dysregulated in FMS patients became relatively normalized by the administration of propranolol. The explanation for this could be a complex reorganization in the central nervous system and/or possible effects on the immune system via adrenergic receptors on leukocytes, or a simple action on sensory neurons that control sympathetic reflexes and muscle pain. The complex CNS and immune explanations seem improbable because most TMD and FMS patients reported a reduction in clinical pain by propranolol (and not by placebo; see Figure 11.11) that was almost immediate (within minutes), without time for reorganization of central sympathetic systems or alterations in immune function.

FIGURE 11.11
Propranolol reduces total body pain. (Reprinted with permission from Light KC, Bragdon EE, Grewen KM, Brownley KA. 2009. Adrenergic dysregulation and pain with and without acute beta-blockade in women with fibromyalgia and temporomandibular disorder. (more...)
The simple explanation was that there are β adrenergic receptors not only on vascular smooth muscle in skeletal muscles, but also on the muscle sensory neurons whose activation signals muscle pain and triggers sympathetic reflexes. To fully explain these phenomena, putative adrenergic receptors on muscle sensory neurons would have to be considerably upregulated in TMD and FMS. A recent case report showing that CFS symptoms could be greatly reduced with propranolol supports this possibility (Wyller et al. 2007). However, there has been no previous demonstration of β adrenergic receptors on muscle sensory neurons.
11.14. BACK TO THE MOUSE TO FIGURE OUT HOW ADRENERGIC RECEPTORS COULD INCREASE MUSCLE PAIN AND FATIGUE
Although paradoxical, alpha-adrenergic receptors have been localized on sensory afferent neurons. Alpha-adrenergic receptors (both α1 and α2) have been shown to upregulate on nociceptive sensory neurons in animal models of neuropathic pain and inflammation, resulting in enhanced nociceptive effects of cutaneous stimulation (Birder and Perl 1999; Lee et al. 1999; O’Halloran and Perl 1997; Sato and Perl 1991). However, alpha-adrenergic receptors on afferent nerves in the spinal cord appear to inhibit pain. In fact, intrathecal treatment with clonidine, an alpha-adrenergic agonist, is often used to reduce various types of pain in patients (Boyd 2001; Brown et al. 2004; Sites et al. 2003).
Only a few reports suggest that beta-adrenergic receptors play a role in enhanced pain following injury (Khasar et al. 1999), and these receptors have not been found on muscle sensory neurons. Given the compelling results from the human experiments, we examined mouse DRG using PCR to determine if beta-adrenergic receptors were present. We also induced gastrocnemius muscle inflammation and used quantitative real-time PCR to determine if beta-adrenergic receptors were upregulated. Finding adrenergic β1 and β2 receptor mRNA present on mouse lumbar DRG neurons, we further observed β2 receptor mRNA is upregulated 24 hours after carrageenan injection into the gastrocnemius muscle (see Figure 11.12), and this upregulation lasted at least 8 days.
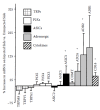
FIGURE 11.12
Twenty-four hour percent increase in mRNA caused by carrageenan-induced muscle inflammation. Note significant increases in adrenergic receptors (n = 8 mice).
To determine if β2 receptor mRNA was upregulated more in neurons innervating the carrageenan-injected muscle, we harvested DRG neurons labeled with DiI from the carrageenan-injected muscle, and compared them with DiI-labeled neurons from the contralateral side, also injected with DiI but not carrageenan-inflamed (Figure 11.13). This procedure revealed that β2 adrenergic mRNA was much higher in DiI-labeled neurons from the carrageenan-inflamed muscle than in muscle-innervating DRG neurons from the contralateral side.

FIGURE 11.13
Quantitative mRNA from DiI-labeled DRG neurons from gastrocnemius muscle injected with 3% carrageenan 24 hours before harvesting neurons indicates that most of the (32 adrenergic receptor increases were in DRG neurons in the inflamed muscle. 100 DRGs—No (more...)
Jon Levine (University of California, San Francisco) was sufficiently intrigued by our animal and human observations to evaluate possible behavioral effects of (β-adrenergic upregulation in rats with the gastrocnemius muscles inflamed by injection of carrageenan, as we had done in mice. He tested sensitivity of the inflamed muscle 1 hour, 24 hours, and 7 days after inflammation. He also tested the muscle sensitivity after injection of propranolol at doses comparable to those used in the human studies described above, and showed (Figure 11.14) that propranolol had no effect 1 hour after induction of inflammation. However, the threshold for a withdrawal response was greatly increased (indicating pain reduction) by propranolol 24 hours after induction of inflammation—when mRNA for beta-adrenergic receptors was increased in DRG neurons.
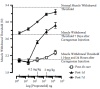
FIGURE 11.14
Effects of propranolol on inflammation-induced muscle pain in male rats. Carr (1% i/m) versus propranolol DRC (n = 6 rats). (Courtesy of Dr. Olayinka Dina and Jon Levine, UCSF.)
Propranolol increased withdrawal threshold an even greater percentage 7 days after inflammation—a time at which the only detectable receptors significantly increased in mouse DRGs were β adrenergic receptors. Dr. Levine has also shown that stress via the hypothalamic-pituitary-adrenal axis (HPA) in combination with sympathetic activation can cause a switch in the adrenergic-signaling pathway causing hyperalgesia in rats. Thus, it appears that β adrenergic receptors on DRG sensory neurons contribute to enhanced sensitivity of inflamed muscle. Whether or not beta-adrenergic receptors play a role in the signal for enhanced sensations of muscle fatigue is unknown, but this seems likely. The same situation may apply to enhanced muscle pain and sensory muscle fatigue in patients with a variety of conditions, including FMS and CFS. Understanding the regulation of beta-adrenergic receptors in muscle sensory neurons, as well as how this regulation interacts with the activity of ASICs, P2X, and TRPV1 receptors in signaling muscle fatigue and pain, may greatly enhance our ability to manage a number of muscle pain and sensory muscle fatigue conditions.
11.15. FUTURE RESEARCH DIRECTIONS
Both CFS and FMS are difficult to diagnose and treat due to a deficiency of objective biomarkers for diagnosis or for quantifying effects of treatment on the primary symptoms in these syndromes: muscle pain and sensations of muscle fatigue. The basis for a medically and legally acceptable objective test might be developed from the leukocyte gene expression changes after exercise that we observed in our patient groups. With these assays, post-exercise blood tests could provide objective validation of a prolonged and severe sensory muscle fatigue and muscle pain state. Combined with assays for other genes modified by other fatiguing and muscle pain-causing diseases, mechanisms of CFS and FMS might be distinguished from other sources of fatigue and pain. Observations on how gene modulation evoked by exercise is altered by treatment may also provide further understanding of CFS and FMS and help guide more effective treatments of these syndromes.
Using these mRNA tests in a prospective study on a population of subjects, some of whom develop CFS and/or FMS, may provide a test for those susceptible to developing these conditions, and could provide information necessary for prevention ofCFSandFMS.
The experiments described here also suggest alternative research directions that could increase understanding of several functional disorders. Some of the genes dys-regulated in CFS and FMS have been implicated in irritable bowel syndrome (IBS), specifically, ASICs, TRPV1, adrenergic (β2, IL10, and TLR4 receptors. The concept that some of these may form receptor complexes that interact to create functional units may be helpful in understanding this syndrome. Similarly, multiple chemical sensitivity might benefit from understanding the receptor complexes that can detect a variety of different chemical signals.
The concept of receptor complexes might also apply to the elusive molecular nature of mechanical nociceptors activated by noxious mechanical stimuli. Identification of these critical molecules could greatly enhance treatment of a variety of pain conditions and syndromes. One particular consideration is that understanding the interaction between the mechanoreceptors and metaboreceptors of Group III and IV muscle sensory neurons might assist description and treatment of restless leg syndrome (RLS). This enigmatic syndrome is characterized by “unpleasant sensations in the legs and an uncontrollable urge to move when at rest in an effort to relieve these feelings” (NINDS 2001). While considered a central neurological problem at present (much like CFS and FMS), the symptoms (the sensation of mechanical stimulation inside the legs) and their alleviation by movement (increasing blood flow to eliminate metabolites) can be explained by activation of muscle Group III, non-nociceptive mechanoreceptors that are also affected by contraction-produced muscle metabolites. Further study of Group III and IV muscle afferent involvement in RLS deserves serious investigation.
Finally, does fatigue constitute a specific sensory system? While our initial efforts indicate that specific receptors may exist, much work needs to be done on characterizing the receptors for fatigue in skeletal muscle, even more on the very important receptors for fatigue in cardiac muscle, and still more on the fatigue receptors in the brain and spinal cord, as well as defining the spinal cord, brain stem, thalamic, and cortical regions that process the sensory aspect of fatigue. With a specific stimulus (the “correct” metabolite “soup” to specifically activate these receptors), brain regions integrating this phenomenon can be identified. It is even possible that the motor regions that receive this signal and are responsible for the noncognitive shutdown of motor command that causes failure of voluntary muscle contraction can be identified by using the knowledge gained by studying the afferent system. It is likely that these motor regions hold a clue to the causes of somatoform disorders that can cause paralysis of specific cognitively controlled movements in some patients.
As with any fertile research area there remain many questions, but we have the tools, and some of the concepts, necessary to begin to investigate the most prevalent symptoms that bring patients to the clinics around the world: fatigue and pain.
ACKNOWLEDGMENTS
Research by the authors reported here was supported by NIH grant R21 NS057821 from NINDS and NIAMS, with additional ancillary support from NIH R21 AT0002209 from NCAM, a catalyst grant from the University of Utah Health Sciences Center, a synergy grant from the University of Utah, and a grant from the Deptartment of Anesthesiology, University of Utah.
The authors would also like to acknowledge the support of the University of Utah Health Sciences Center Genomics and Imaging Cores. Special thanks to Dr. Lucinda Bateman for help with patient referrals. Thanks to Drs. Rajan Radhakrishnan and Jesse Zhang for their help on projects described in this chapter. Also, our thanks to undergraduate students supported by UROP and BIOURP programs including Benjamin Jensen, Cody Larson, Tania Michael, Clay Peterson, Nick Daskalas, Sean Gowen, Shane Hawthorne, Sonia Sarfraz, Tuyet Nguyen, and graduate students from the Exercise and Sport Science Department including K.L. Fitschen, Luke Wendt, and Tim Vanhaitsma.
REFERENCES
- Adreani CM, Hill JM, Kaufman MP. Responses of group III and IV muscle afferents to dynamic exercise. J Appl Physiol. 1997;82:1811–1817. [PubMed: 9173945]
- Adreani CM, Kaufman MP. Effect of arterial occlusion on responses of group III and IV afferents to dynamic exercise. J Appl Physiol. 1998;84:1827–1833. [PubMed: 9609773]
- Alam M, Smirk FH. Observations in man upon a blood pressure raising reflex arising from the voluntary muscles. J Physiol. 1937;89:372–383. [PMC free article: PMC1395054] [PubMed: 16994867]
- Ambalavanar R, Moritani M, Moutanni A, Gangula P, Yallampalli C, Dessem D. Deep tissue inflammation upregulates neuropeptides and evokes nociceptive behaviors which are modulated by a neuropeptide antagonist. Pain. 2006;120:53–68. [PubMed: 16359792]
- Ambalavanar R, Yallampalli C, Yallampalli U, Dessem D. Injection of adjuvant but not acidic saline into craniofacial muscle evokes nociceptive behaviors and neuropeptide expression. Neuroscience. 2007;149:650–659. [PMC free article: PMC2196131] [PubMed: 17928159]
- Baekken PM, Skorpen F, Stordal E, Zwart JA, Hagen K. Depression and anxiety in relation to catechol-O-methyltransferase Val158Met genotype in the general population: the Nord-Trondelag Health Study (HUNT). BMC Psychiatry. 2008;8:48. [PMC free article: PMC2453123] [PubMed: 18578865]
- Bellinger AM, Mongillo M, Marks AR. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest. 2008;118:445–453. [PMC free article: PMC2214709] [PubMed: 18246195]
- Birder LA, Perl ER. Expression of alpha2-adrenergic receptors in rat primary afferent neurones after peripheral nerve injury or inflammation. J Physiol. 1999;515(Pt 2):533–542. [PMC free article: PMC2269161] [PubMed: 10050019]
- Boyd RE. Alpha2-adrenergic receptor agonists as analgesics. Curr Top Med Chem. 2001;1:193–197. [PubMed: 11895135]
- Brooks JC, Zambreanu L, Godinez A, Craig AD, Tracey I. Somatotopic organisation of the human insula to painful heat studied with high resolution functional imaging. Neuroimage. 2005;27:201–209. [PubMed: 15921935]
- Brown DR, Hofer RE, Patterson DE, Fronapfel PJ, Maxson PM, Narr BJ, Eisenach JH, Blute ML, Schroeder DR, Warner DO. Intrathecal anesthesia and recovery from radical prostatectomy: a prospective, randomized, controlled trial. Anesthesiology. 2004;100:926–934. [PubMed: 15087629]
- Caseras X, Mataix-Cols D, Rimes KA, Giampietro V, Brammer M, Zelaya F, Chalder T, Godfrey E. The neural correlates of fatigue: an exploratory imaginal fatigue provocation study in chronic fatigue syndrome. Psychol Med. 2008;38:941–951. [PubMed: 18447963]
- Chacur M, Lambertz D, Hoheisel U, Mense S. Role of spinal microglia in myositis-induced central sensitisation: An immunohistochemical and behavioural study in rats. Eur J Pain. 2008 [PubMed: 19095475]
- Cheung K, Hume P, Maxwell L. Delayed onset muscle soreness: treatment strategies and performance factors. Sports Med. 2003;33:145–164. [PubMed: 12617692]
- Christianson JA, McIlwrath SL, Koerber HR, Davis BM. Transient receptor potential vanilloid 1-immunopositive neurons in the mouse are more prevalent within colon affer-ents compared to skin and muscle afferents. Neuroscience. 2006;140:247–257. [PubMed: 16564640]
- Connor M, Naves LA, McCleskey EW. Contrasting phenotypes of putative proprioceptive and nociceptive trigeminal neurons innervating jaw muscle in rat. Mol Pain. 2005;1 [PMC free article: PMC1283980] [PubMed: 16242047]
- Cook DB, Lange G, Ciccone DS, Liu WC, Steffener J, Natelson BH. Functional imaging of pain in patients with primary fibromyalgia. J Rheumatol. 2004;31:364–378. [PubMed: 14760810]
- Cook DB, O’Connor PJ, Lange G, Steffener J. Functional neuroimaging correlates of mental fatigue induced by cognition among chronic fatigue syndrome patients and controls. Neuroimage. 2007;36:108–122. [PubMed: 17408973]
- Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha XM, Price M, Schnizler MK, Wemmie JA. Targeting ASIC la reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry. 2007;62(10):1140–1148. [PubMed: 17662962]
- Craig AD. Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci. 2003;26:1–30. [PubMed: 12651967]
- Cristino L, Starowicz K, De Petrocellis L, Morishita J, Ueda N, Guglielmotti V, Di M. V. Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience. 2008;151:955–968. [PubMed: 18248904]
- Davis JM, Bailey SP. Possible mechanisms of central nervous system fatigue during exercise. Med Sci Sports Exerc. 1997;29:45–57. [PubMed: 9000155]
- Di M, V, Gobbi G, Szallasi A. Brain TRPV1: a depressing TR(i)P down memory lane? Trends Pharmacol Sci. 2008;29:594–600. [PubMed: 18947889]
- Diatchenko L, Anderson AD, Slade GD, Fillingim RB, Shabalina SA, Higgins TJ, Sama S, et al. Three major haplotypes of the beta 2 adrenergic receptor define psychological profile, blood pressure, and the risk for development of a common musculoskeletal pain disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141:449–462. [PMC free article: PMC2570772] [PubMed: 16741943]
- Drust B, Rasmussen P, Mohr M, Nielsen B, Nybo L. Elevations in core and muscle temperature impairs repeated sprint performance. Acta Physiol Scand. 2005;183:181–190. [PubMed: 15676059]
- Falk K, Swedberg K, Gaston-Johansson F, Ekman I. Fatigue is a prevalent and severe symptom associated with uncertainty and sense of coherence in patients with chronic heart failure. Eur J Cardiovasc Nurs. 2006;6(2):99–104. [PubMed: 16831569]
- Funke B, Malhotra AK, Finn CT, Plocik AM, Lake SL, Lencz T, DeRosse P, Kane JM, Kucherlapati R. COMT genetic variation confers risk for psychotic and affective disorders: a case control study. Behav Brain Funct. 2005;1:19. [PMC free article: PMC1282571] [PubMed: 16232322]
- Geisser ME, Strader DC, Petzke F, Gracely RH, Clauw DJ, Williams DA. Comorbid somatic symptoms and functional status in patients with fibromyalgia and chronic fatigue syndrome: sensory amplification as a common mechanism. Psychosomatics. 2008;49:235–242. [PubMed: 18448779]
- Gibson A, Baden D, Lambert MI, Lambert EV, Harley YXR, Hampson D, Russell VA, Noakes TD. The conscious perception of the sensation of fatigue. Sports Med. 2003;33:167–176. [PubMed: 12656638]
- Gibson W, Arendt-Nielsen L, Taguchi T, Mizumura K, Graven-Nielsen T. Increased pain from muscle fascia following eccentric exercise: animal and human findings. Exp Brain Res. 2009 [PubMed: 19156402]
- Goertzel BN, Pennachin C, de Souza CL, Gurbaxani B, Maloney EM, Jones JF. Combinations of single nucleotide polymorphisms in neuroendocrine effector and receptor genes predict chronic fatigue syndrome. Pharmacogenomics. 2006;7:475–483. [PubMed: 16610957]
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum. 2002;46:1333–1343. [PubMed: 12115241]
- Hayes SG, Kindig AE, Kaufman MP. Cyclooxygenase blockade attenuates responses of group III and IV muscle afferents to dynamic exercise in cats. Am J Physiol Heart Circ Physiol. 2006;290:H2239–H2246. [PubMed: 16399856]
- Hildebrand MS, de Silva MG, Klockars T, Rose E, Price M, Smith RJ, McGuirt WT, Christopoulos H, Petit C, Dahl HH. Characterisation of DRASIC in the mouse inner ear. Hear Res. 2004;190:149–160. [PubMed: 15051137]
- Hoheisel U, Unger T, Mense S. Excitatory and modulatory effects of inflammatory cytokines and neurotrophins on mechanosensitive group IV muscle afferents in the rat. Pain. 2005;114:168–176. [PubMed: 15733642]
- Holmes GP, Kaplan JE, Gantz NM, Komaroff AL, Schonberger LB, Straus SE, Jones JF, Dubois RE, Cunningham-Rundles C, Pahwa S. Chronic fatigue syndrome: a working case definition. Ann Intern Med. 1988;108:387–389. [PubMed: 2829679]
- Immke DC, McCleskey EW. ASIC3: a lactic acid sensor for cardiac pain. ScientificWorldJournal. 2001a;1:510–512. [PMC free article: PMC6084709] [PubMed: 12805843]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001b;4:869–870. [PubMed: 11528414]
- Immke DC, McCleskey EW. Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron. 2003;37:75–84. [PubMed: 12526774]
- Jones RC III, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–10989. [PMC free article: PMC6725875] [PubMed: 16306411]
- Kaufman MP. Has the phoenix risen? J Physiol. 2003;548:666. [PMC free article: PMC2342896] [PubMed: 12640003]
- Kaufman MP, Hayes SG. The exercise pressor reflex. Clin Auton Res. 2002;12:429–439. [PubMed: 12598947]
- Khan MH, Sinoway LI. Muscle reflex control of sympathetic nerve activity in heart failure: the role of exercise conditioning. Heart Fail Rev. 2000;5:87–100. [PubMed: 16228918]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999;81:1104–1112. [PubMed: 10085337]
- Kniffki KD, Mense S, Schmidt RF. Responses of group IV afferent units from skeletal muscle to stretch, contraction and chemical stimulation. Exp Brain Res. 1978;31:511–522. [PubMed: 658178]
- Kniffki KD, Mense S, Schmidt RF. Muscle receptors with fine afferent fibers which may evoke circulatory reflexes. Circ Res. 1981;48:1125–131. [PubMed: 7014023]
- Kruger K, Lechtermann A, Fobker M, Volker K, Mooren FC. Exercise-induced redistribution of T lymphocytes is regulated by adrenergic mechanisms. Brain Behav Immun. 2008;22:324–338. [PubMed: 17910910]
- Kruger L, Silverman JD, Mantyh PW, Sternini C, Brecha NC. Peripheral patterns of cal-citonin-gene-related peptide general somatic sensory innervation: cutaneous and deep terminations. J Comp Neurol. 1989;280:291–302. [PubMed: 2784448]
- Lee DH, Liu X, Kim HT, Chung K, Chung JM. Receptor subtype mediating the adrenergic sensitivity of pain behavior and ectopic discharges in neuropathic Lewis rats. J Neurophysiol. 1999;81:2226–2233. [PubMed: 10322061]
- Leffler A, Monter B, Koltzenburg M. The role of the capsaicin receptor TRPV1 and acid-sensing ion channels (ASICS) in proton sensitivity of subpopulations of primary nociceptive neurons in rats and mice. Neuroscience. 2006;139:699–709. [PubMed: 16515841]
- Li Y, Ji A, Weihe E, Schafer MK. Cell-specific expression and lipopolysaccharide-induced regulation of tumor necrosis factor alpha (TNF alpha) and TNF receptors in rat dorsal root ganglion. J Neurosci. 2004;24:9623–9631. [PMC free article: PMC6730137] [PubMed: 15509749]
- Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J. Dorsal root ganglion neurons innervating skeletal muscle respond to physiological combinations of protons, ATP, and lactate mediated by ASIC, P2X, and TRPV1. J Neurophysiol. 2008;100:1184–1201. [PMC free article: PMC6195653] [PubMed: 18509077]
- Light AR, White AT, Hughen RW, Light KC. Moderate exercise increases expression for sensory, adrenergic, and immune genes in chronic fatigue syndrome patients, but not in normal subjects. J Pain. epub July 30. [PMC free article: PMC2757484] [PubMed: 19647494]
- Light KC, Bragdon EE, Grewen KM, Brownley KA. Adrenergic dysregulation and pain with and without acute beta-blockade in women with fibromyalgia and temporomandibular disorder. J Pain. 2009b;10:452–552. [PMC free article: PMC2700184] [PubMed: 19411061]
- Mamet J, Lazdunski M, Voilley N. How nerve growth factor drives physiological and inflammatory expressions of acid-sensing ion channel 3 in sensory neurons. J Biol Chem. 2003;278:48907–48913. [PubMed: 14522957]
- Mathew SJ, Mao X, Keegan KA, Levine SM, Smith EL, Heier LA, Otcheretko V, Coplan JD, Shungu DC. Ventricular cerebrospinal fluid lactate is increased in chronic fatigue syndrome compared with generalized anxiety disorder: an in vivo 3.0 T (1)H MRS imaging study. NMR Biomed. 2009;22(3):251–258. [PubMed: 18942064]
- Mense S. Algesic agents exciting muscle nociceptors. Exp Brain Res. 2009;196:89–100. [PubMed: 19139871]
- Mense S, Meyer H. Different types of slowly conducting afferent units in cat skeletal muscle and tendon. J Physiol (Lond). 1985;363:403–417. [PMC free article: PMC1192937] [PubMed: 4020704]
- Micevych PE, Kruger L. The status of calcitonin gene-related peptide as an effector peptide. Ann NY Acad Sci. 1992;657:379–396. [PubMed: 1322091]
- Molliver DC, Immke DC, Fierro L, Pare M, Rice FL, McCleskey EW. ASIC3, an acid-sensing ion channel, is expressed in metaboreceptive sensory neurons. Mol Pain. 2005;1:35. [PMC free article: PMC1308857] [PubMed: 16305749]
- Mosso A. Fatigue. 1904
- Müller J. Koblenz, Germany:: Holscher J; 1840. Handbuch der Physiologie des Menschen.
- Naves LA, McCleskey EW. An acid-sensing ion channel that detects ischemic pain. Braz J Med Biol Res. 2005;38:1561–1569. [PubMed: 16258623]
- NINDS. 2001. Restless Legs Syndrome Fact Sheet. NIH Publication No 01-4847. www
.ninds.nih.gov/disorders /restless_legs /detail_restless_legs.htm - Noakes TD. The central governor model of exercise regulation applied to the marathon. Sports Med. 2007;37:374–377. [PubMed: 17465612]
- Noakes TD, St Clair GA, Lambert EV. From catastrophe to complexity: a novel model of integrative central neural regulation of effort and fatigue during exercise in humans: summary and conclusions. Br J Sports Med. 2005;39:120–124. [PMC free article: PMC1725112] [PubMed: 15665213]
- O’Halloran KD, Perl ER. Effects of partial nerve injury on the responses of C-fiber polymodal nociceptors to adrenergic agonists. Brain Res. 1997;759:233–240. [PubMed: 9221942]
- Ohara K, Nagai M, Suzuki Y, Ohara K. Low activity allele of catechol-o-methyltrans- ferase gene and Japanese unipolar depression. Neuroreport. 1998;9:1305–1308. [PubMed: 9631418]
- Page AJ, Brierley SM, Martin CM, Price MP, Symonds E, Butler R, Wemmie JA, Blackshaw LA. Different contributions of ASIC channels la, 2, and 3 in gastrointestinal mechanosensory function. Gut. 2005;54:1408–1415. [PMC free article: PMC1774697] [PubMed: 15987792]
- Pall ML, Anderson JH. The vanilloid receptor as a putative target of diverse chemicals in multiple chemical sensitivity. Arch Environ Health. 2004;59:363–375. [PubMed: 16241041]
- Papka RE, McNeill DL. Light- and electron-microscopic study of synaptic connections in the paracervical ganglion of the female rat: special reference to calcitonin gene-related peptide-, galanin- and tachykinin (substance P and neurokinin A)-immunoreactive nerve fibers and terminals. Cell Tissue Res. 1993;271:417–428. [PubMed: 7682477]
- Peng BG, Ahmad S, Chen S, Chen P, Price MP, Lin X. Acid-sensing ion channel 2 contributes a major component to acid-evoked excitatory responses in spiral ganglion neurons and plays a role in noise susceptibility of mice. J Neurosci. 2004;24:10167–10175. [PMC free article: PMC6730178] [PubMed: 15537887]
- Perl ER. Is pain a specific sensation. J Psychiatr Res. 1971;8:273–287. [PubMed: 4939377]
- Perl ER. Getting a line on pain: is it mediated by dedicated pathways? Nat Neurosci. 1998;1:177–178. [PubMed: 10195139]
- Perl ER. Ideas about pain, a historical view. Nat Rev Neurosci. 2007;8:71–80. [PubMed: 17180164]
- Reeves WC, Jones JF, Maloney E, Heim C, Hoaglin DC, Boneva RS, Morrissey M, Devlin R. Prevalence of chronic fatigue syndrome in metropolitan, urban, and rural Georgia. Popul Health Metr. 2007;5:5. [PMC free article: PMC1904178] [PubMed: 17559660]
- Reinohl J, Hoheisel U, Unger T, Mense S. Adenosine triphosphate as a stimulant for nociceptive and non-nociceptive muscle group IV receptors in the rat. Neurosci Lett. 2003;338:25–28. [PubMed: 12565132]
- Rybicki KJ, Waldrop TG, Kaufman MP. Increasing gracilis muscle interstitial potassium concentrations stimulate group III and IV afferents. JAppl Physiol. 1985;58:936–941. [PubMed: 2984167]
- Sato J, Perl ER. Adrenergic excitation of cutaneous pain receptors induced by peripheral nerve injury. Science. 1991;251:1608–1610. [PubMed: 2011742]
- Schmaling KB, Lewis DH, Fiedelak JI, Mahurin R, Buchwald DS. Single-photon emission computerized tomography and neurocognitive function in patients with chronic fatigue syndrome. Psychosom Med. 2003;65:129–136. [PubMed: 12554824]
- Silverman JD, Kruger L. Calcitonin-gene-related-peptide-immunoreactive innervation of the rat head with emphasis on specialized sensory structures. J Comp Neurol. 1989;280:303–330. [PubMed: 2784449]
- Sites BD, Beach M, Biggs R, Rohan C, Wiley C, Rassias A, Gregory J, Fanciullo G. Intrathecal clonidine added to a bupivacaine-morphine spinal anesthetic improves postoperative analgesia for total knee arthroplasty. Anesth Analg. 2003;96:1083–8. table. [PubMed: 12651665]
- St Clair GA, Noakes TD. Evidence for complex system integration and dynamic neural regulation of skeletal muscle recruitment during exercise in humans. Br J Sports Med. 2004;38:797–806. [PMC free article: PMC1724966] [PubMed: 15562183]
- Staud R, Cannon RC, Mauderli AP, Robinson ME, Price DD, Vierck CJ Jr. Temporal summation of pain from mechanical stimulation of muscle tissue in normal controls and subjects with fibromyalgia syndrome. Pain. 2003;102:87–95. [PubMed: 12620600]
- Sugiura T, Bielefeldt K, Gebhart GR. Mouse colon sensory neurons detect extracellular acidosis via TRPV1. Am J Physiol Cell Physiol. 2007;292:C1768–C1774. [PubMed: 17251322]
- Sugiura T, Kasai M, Katsuya H, Mizumura K. Thermal properties of acid-induced depolarization in cultured rat small primary afferent neurons. Neurosci Lett. 2003;350:109–112. [PubMed: 12972165]
- Takahashi K, Taguchi T, Itoh K, Okada K, Kawakita K, Mizumura K. Influence of surface anesthesia on the pressure pain threshold measured with different-sized probes. Somatosens Mot Res. 2005;22:299–305. [PubMed: 16503582]
- Toth A, Boczan J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135:162–168. [PubMed: 15857679]
- Tsai SJ, Gau YT, Hong CJ, Liou YJ, Yu YW, Chen TJ. Sexually dimorphic effect of catechol-O-methyltransferase vall58met polymorphism on clinical response to fluoxetine in major depressive patients. J Affect Disord. 2009;113:183–187. [PubMed: 18533273]
- Vierck CJ Jr. Mechanisms underlying development of spatially distributed chronic pain (fibromyalgia). Pain. 2006;124:242–263. [PubMed: 16842915]
- Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21:8026–8033. [PMC free article: PMC6763876] [PubMed: 11588175]
- Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997a;272:20975–20978. [PubMed: 9261094]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997b;386:173–177. [PubMed: 9062189]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH Jr., Welsh MJ. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23:5496–5502. [PMC free article: PMC6741257] [PubMed: 12843249]
- Wemmie JA, Coryell MW, Askwith CC, Lamani E, Leonard AS, Sigmund CD, Welsh MJ. Overexpression of acid-sensing ion channel la in transgenic mice increases acquired fear-related behavior. Proc Natl Acad Sci USA. 2004;101:3621–3626. [PMC free article: PMC373512] [PubMed: 14988500]
- Williamson JW, Nobrega AC, McColl R, Mathews D, Winchester P, Friberg L, Mitchell JH. Activation of the insular cortex during dynamic exercise in humans. J Physiol. 1997;503(Pt 2):277–283. [PMC free article: PMC1159862] [PubMed: 9306272]
- Wilson LB, Andrew D, Craig AD. Activation of spinobulbar lamina I neurons by static muscle contraction. J Neurophysiol. 2002;87:1641–1645. [PubMed: 11877534]
- Wright-Browne V, Schnee AM, Jenkins MA, Thall PF, Aggarwal BB, Talpaz M, Estrov Z. Serum cytokine levels in infectious mononucleosis at diagnosis and convales cence. Leuk Lymphoma. 1998;30:583–589. [PubMed: 9711920]
- Wyller VB, Thaulow E, Amlie JP. Treatment of chronic fatigue and orthostatic intolerance with propranolol. JPediatr. 2007;150:654–655. [PubMed: 17517256]
- Yagi J, Wenk HN, Naves LA, McCleskey EW. Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ Res. 2006;99:501–509. [PubMed: 16873722]
- Yoshiuchi K, Farkas J, Natelson BH. Patients with chronic fatigue syndrome have reduced absolute cortical blood flow. Clin Physiol Fund Imaging. 2006;26:83–86. [PubMed: 16494597]
- INTRODUCTION
- DO SKELETAL MUSCLES HAVE TWO UNIQUE TYPES OF SENSORY RECEPTORS THAT DETECT METABOLITES: ONE TYPE NOCICEPTIVE (CAPABLE OF SIGNALING PAIN), THE OTHER ERGORECEPTIVE (CAPABLE OF DETECTING MUSCLE WORK)?
- MOLECULAR RECEPTORS THAT ARE ACTIVATED BY METABOLITES PRODUCED BY MUSCLE CONTRACTION
- THE NEED FOR TRANSLATIONAL BRIDGES TO FIBROMYALGIA AND CHRONIC FATIGUE SYNDROME
- ARE THESE MOLECULAR RECEPTORS RESPONSIBLE FOR SENSORY MUSCLE FATIGUE AND MUSCLE PAIN?
- TRANSLATION FROM MOUSE TO HUMAN
- MOLECULAR RECEPTORS INVOLVED IN CHRONIC FATIGUE SYNDROME
- DYSREGULATED MRNAS MAY ALSO EXPLAIN COMMON CO-MORBIDITIES OF CFS
- CAN ACUTE FATIGUE LEAD TO CHRONIC FATIGUE?
- COMMON CAUSES OF ACUTE FATIGUE MIGHT CAUSE UP-REGULATION OF THE mRNA FOR THE MOLECULAR RECEPTORS SUGGESTED TO MEDIATE SENSORY MUSCLE FATIGUE AND PAIN
- MENTAL FATIGUE
- SYMPATHETIC DYSREGULATION CONTRIBUTES TO ENHANCED SENSORY FATIGUE
- ADRENERGIC RECEPTORS ON SENSORY NEURONS?
- BACK TO THE MOUSE TO FIGURE OUT HOW ADRENERGIC RECEPTORS COULD INCREASE MUSCLE PAIN AND FATIGUE
- FUTURE RESEARCH DIRECTIONS
- ACKNOWLEDGMENTS
- REFERENCES
- Overlapping conditions among patients with chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder.[Arch Intern Med. 2000]Overlapping conditions among patients with chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder.Aaron LA, Burke MM, Buchwald D. Arch Intern Med. 2000 Jan 24; 160(2):221-7.
- Review Physiological accompaniments of human muscle fatigue.[Aust Paediatr J. 1988]Review Physiological accompaniments of human muscle fatigue.Gandevia SC. Aust Paediatr J. 1988; 24 Suppl 1:104-8.
- Moderate exercise increases expression for sensory, adrenergic, and immune genes in chronic fatigue syndrome patients but not in normal subjects.[J Pain. 2009]Moderate exercise increases expression for sensory, adrenergic, and immune genes in chronic fatigue syndrome patients but not in normal subjects.Light AR, White AT, Hughen RW, Light KC. J Pain. 2009 Oct; 10(10):1099-112. Epub 2009 Jul 31.
- Review Pathophysiology of Mild TBI: Implications for Altered Signaling Pathways.[Brain Neurotrauma: Molecular, ...]Review Pathophysiology of Mild TBI: Implications for Altered Signaling Pathways.Laskowski RA, Creed JA, Raghupathi R. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. 2015
- Review Exercise therapy for chronic fatigue syndrome.[Cochrane Database Syst Rev. 2017]Review Exercise therapy for chronic fatigue syndrome.Larun L, Brurberg KG, Odgaard-Jensen J, Price JR. Cochrane Database Syst Rev. 2017 Apr 25; 4(4):CD003200. Epub 2017 Apr 25.
- Myalgia and Fatigue - Translational Pain ResearchMyalgia and Fatigue - Translational Pain Research
Your browsing activity is empty.
Activity recording is turned off.
See more...