A new version of this title is available
Summary
- Duchenne muscular dystrophy (DMD) is a rare and severe disorder that affects primarily young boys. It begins with progressive muscle weakness that evolves to loss of ambulation and further progresses to early morbidity and mortality.
- DMD is caused by a mutation of the dystrophin gene that results in a lack of dystrophin, a protein that is necessary for muscle cell function.
- The mainstays of current therapy to treat DMD are corticosteroids and assistive devices.
- This bulletin focuses on new and emerging drugs for DMD with completed phase IIb or phase III trials and includes ataluren (nonsense mutation suppression), eteplirsen (exon 51 skipping), and idebenone (adenosine triphosphate [ATP] modulation). Ezutromid (utrophin modulation) and givinostat (histone deacetylase inhibition) are in early stages of development.
- The evidence reviewed showed that, while these drugs increased the production of new dystrophin, they generally had limited impact on ambulation and physical function.
Background
DMD is an X-linked genetic disorder and is the most common form of muscular dystrophy in childhood, affecting one in 3,500 to 6,000 newborn males.1,2 There are approximately 50,000 cases of DMD worldwide.3 In 2015, it was estimated there were 794 DMD patients in Canada.4 The disease is caused by mutations of the DMD gene that codes for dystrophin, a protein that protects the integrity of muscle cells.5–7
The loss of dystrophin in patients with DMD disrupts the muscle membrane and fibres. This leads to progressive muscle weakness, gait disturbance, motor development delay, calf hypertrophy, and elevated creatine kinase levels (a biomarker reflecting muscle damage).6 Most affected children first present with symptoms between the ages of three and five years. These symptoms gradually worsen with time, eventually leading to loss of ambulation and the need to use a wheelchair by early adolescence.8 Boys with DMD may also have intellectual disability, speech delay, and cognitive impairment.6,8 Further progression of the disease leads to respiratory insufficiency and cardiomyopathy, which is present in almost all patients by 18 years of age.8 Death typically occurs before the third decade of life due to cardiorespiratory complications.6,8,9 Female carriers of the DMD mutation are largely asymptomatic, although 20% to 30% may present with mild-to-moderate muscle weakness, and are at increased risk for dilated cardiomyopathy.8
There is an unmet medical need for the treatment of DMD, as there is no cure and the standard of care is limited to corticosteroids and assistive devices.
The Technology
There are currently two genetic strategies in development that attempt to re-establish the expression of dystrophin in muscle: suppression of nonsense mutations and exon skipping. Further strategies to compensate for the lack of dystrophin include utrophin upregulation, ATP modulation, and histone deacetylase inhibition.
- Ataluren suppresses nonsense mutations, which enables the production of a modified dystrophin protein.
- Eteplirsen is a third-generation synthetic antisense oligonucleotide (AON) that is designed to induce exon 51 skipping.
- Ezutromid is an utrophin modulator that aims to replace lost dystrophin with utrophin.
- Givinostat is a histone deacetylase inhibitor that may help to rebalance the repair process in muscle by increasing muscle regeneration.
- Idebenone is an ATP production modulator that helps dystrophic muscle cells maintain their cellular energy supply and protects cells from oxidative stress.
Appendix 1 contains further details on their mechanisms of action.
Regulatory Status
On July 31, 2014, the European Commission granted a marketing authorization for ataluren (Translarna; PTC Therapeutics) that is valid throughout the European Union. It is also approved in Iceland, Liechtenstein, Norway, Israel, and South Korea. It is indicated for the treatment of DMD in ambulatory patients five years and older with nonsense mutations.10 In Canada, the manufacturer, PTC Therapeutics, had initially submitted the dossier to Health Canada for review in August 2015; in March 2016, it chose to cancel the submission in order to re-file at a later date with additional data.11
The US FDA approved eteplirsen (AVI 4658, Exondys 51; Sarepta Therapeutics) on September 19, 2016 through the accelerated approval pathway, with the condition that a confirmatory clinical trial be conducted.12 In the US, it is indicated for patients who have confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which is about 13% of the DMD population.13
Ezutromid (SMT C1100; Summit Therapeutics), givinostat (ITF2357; Italfarmaco SpA), and idebenone (CV 2619, Raxone or Catena; Santhera Pharmaceuticals) have not received regulatory approval in any country for the treatment of DMD.
Target Population
Ataluren’s intended use is in patients with confirmed nonsense mutation in the dystrophin gene, representing 10% to 15% of the DMD population.14,15
Eteplirsen is for use in children with confirmed mutation of the DMD gene amenable to exon 51 skipping, which is approximately 13% of the DMD population.16
Idebenone has been studied in DMD patients with declining lung function.17,18
Ezutromid and givinostat have the potential to be used in all DMD patients.19,20
Current Practice
There is currently no cure for DMD. Multidisciplinary management includes assistive devices, rehabilitative management, treatment of complications, and genetic counselling.8 Recent advances in management, as well as pharmacological treatment with corticosteroids, has resulted in survival and quality-of-life improvements.8,21
- The mainstay of treatment for DMD is glucocorticoids. There is evidence of their benefit in improving lung and motor function, stabilizing muscle strength, reducing the risk of scoliosis, and prolonging ambulation.1,6,22 The dosage for oral prednisone is generally 0.75mg/kg daily, although alternate-day and high weekend-dose regimens are also used.22 Deflazacort, a derivative of prednisone, is also used in DMD patients at 0.9 mg/kg/day, although it is available only through the Special Access Programme in Canada.
- Cardiac disease, including left ventricular dysfunction and heart failure, is usually managed with angiotensin-converting enzyme inhibitors and beta blockers.23
- Various methods of non-invasive mechanical ventilation have played a major role in increasing survival in DMD patients from their late teens to their late 20s.18
- Orthopedic interventions and physical therapy are also necessary to reduce contractures, manage scoliosis, and maintain independence.8
Summary of the Evidence
Methods
A literature search was conducted using the following bibliographic databases: MEDLINE, PubMed, Embase, and the Cochrane Library (2016, Issue 11). Grey literature was identified by searching relevant sections of the Grey Matters checklist ( https://www.cadth.ca/grey-matters ). Methodological filters were applied to limit retrieval to randomized controlled trials (RCTs). The search was limited to English-language documents published between Jan. 1, 2011 and Dec. 1, 2016. Regular alerts were established to update the search until March 6, 2017. Conference abstracts published between Jan. 1, 2014 and Dec. 1, 2016 were included in the search results.
Efficacy
Appendix 2 contains the characteristics and results of the included trials. Information on ongoing phase III trials for DMD medications is available in Appendix 3.
Study Design
A total of four RCTs are included as evidence: one phase IIb trial and one phase III trial on ataluren; one phase IIb trial on eteplirsen; and one phase III trial on idebenone. Information on an open-label extension trial on eteplirsen is also included. No published phase IIb or phase III trial information on givinostat or ezutromid was found.
Population
The ataluren and eteplirsen trials enrolled patients between the ages of five and 14 years who were ambulatory at screening; the majority were on corticosteroids. The idebenone study population ranged in age from 10 to 18 years, was largely non-ambulatory, and was not on corticosteroids.
Interventions
Ataluren
In the phase IIb trial on ataluren (n = 174), 57 patients were randomized to placebo, 57 patients were randomized to ataluren 40 mg/kg/day in three divided doses, and 60 patients were randomized to ataluren 80 mg/kg/day in three divided doses for 48 weeks.14 The phase III RCT (n = 230) compared ataluren 40 mg/kg day, given in three divided doses (n = 115), with placebo (n = 115) for 48 weeks.15
Eteplirsen
In Study 201, four patients were randomized to the placebo group, four patients to 30 mg/kg of eteplirsen weekly, and four patients to 50 mg/kg of eteplirsen weekly for 24 weeks.16 Study 202 is an extension trial of Study 201 in which eteplirsen-treated patients continued treatment at the same dose and placebo patients were assigned either dose of the study medication starting at week 25 (called the placebo-delayed group; it was unclear if the assignment was randomized). Study 202 is 212 weeks in duration and is ongoing.16
Idebenone
A total of 66 patients were enrolled in the phase III trial: 32 patients were randomized to idebenone 900 mg orally, given as 300 mg three times daily; 32 patients were randomized to matching placebo; and one patient was allocated to the same treatment as his brother. Study duration was 52 weeks.17
Outcomes Measured in Trials
Ambulation
The outcome measures for trials involving drugs aimed at prolonging ambulation in patients with DMD included the six-minute walk test (6MWT; measured in the ataluren and eteplirsen trials); the North Star Ambulatory Assessment (NSAA; measured in the ataluren phase III trial), and physical functioning (measured in both ataluren trials).
- The 6MWT assesses physical function and endurance by measuring the distance a person is able to walk during a total of six minutes on a hard, flat surface. The primary measurement of the 6MWT is the six-minute walk distance (6MWD). A 30 metre change in distance on the 6MWT has been proposed as the minimal clinically important difference in DMD patients with nonsense mutations.24
- The NSAA, is a functional scale consisting of 17 activities to measure disease progression in ambulatory DMD patients.25 For each of these activities, the NSAA assigns a grade from 0 (unable to achieve activity independently) to 2 (“normal” — able to achieve activity without modification), with a maximum possible score of 34, which may be linearized from 0 to 100.25
Dystrophin Production
The eteplirsen trial measured the change in dystrophin production by performing muscle biopsies to measure dystrophin-positive fibres. The laboratory tests used to quantify dystrophin were immunohistochemical and immunofluorescence analyses, and Western Blot.16
Pulmonary Function
Change in respiratory function was measured as peak expiratory flow as percentage predicted (PEF%p). Patients self-assessed their respiratory function weekly.17
Results
A summary of the results is available in Table 1.
Table 1
Results of Phase IIb and Phase III Clinical Trials
Ambulation
Ataluren
In Study 007, the difference in the mean change from baseline in the 6MWD was 29.7 metres for ataluren 40 mg/kg compared with placebo (P = 0.149). There was no statistically significant difference between ataluren 80 mg/kg and placebo. For ataluren 40 mg/kg compared with placebo, the difference in the mean change from baseline in timed function tests showed an improvement of 2.4 seconds in time to climb stairs (95% confidence interval [CI], −4.8 to 0), an improvement of 1.6 seconds in time to descend four stairs (95% CI, 4.2 to 1.0), and an improvement of 1.4 seconds in time to run or walk 10 metres (95% CI, −3.7 to 0.9).14 In Study 020, there was a difference of 15.4 metres in the mean change from baseline in 6MWD between ataluren 40 mg/kg and placebo (statistical significance not reported).15 There was no statistically significant difference in NSAA mean score between ataluren 40 mg/kg and placebo (P = 0.268).25
Eteplirsen
At 24 weeks, the 30 mg/kg group and the placebo group had worsening ambulation, as measured by the 6MWT, compared with baseline (−128.2 metres and −25.8 metres, respectively); there was no change for the patients who were administered 50 mg/kg of eteplirsen. At 48 weeks, the ambulation of the group receiving 50 mg/kg of eteplirsen improved by 21 metres; whereas with 30 mg/kg of eteplirsen, there was a decline of 153.4 metres on the 6MWT from baseline.16
Dystrophin Production
Eteplirsen
At 24 weeks, the production of dystrophin increased by 22.9% from baseline in patients who were administered 30 mg/kg of eteplirsen. Compared with placebo, this increase was statistically significant (mean difference 27%, P < 0.002).16 In the open-label extension trial, at 48 weeks, the production of dystrophin increased by 51.7% from baseline in the patients who were administered 30 mg/kg of eteplirsen.16
Pulmonary Function
Idebenone
In the DELOS trial, both the treated group and the placebo group had worsening respiratory function at week 52. The placebo group had a decline in PEF%p of 8.84% compared with a decline of 2.57% in the idebenone-treated group, resulting in a difference of 6.27% (95% CI, 0.61 to 11.93; P = 0.031).
Study Limitations
The eteplirsen trial (Study 201) had 12 participants, thereby limiting its power and generalizability.
The production of new dystrophin as a surrogate biomarker has been used to grant conditional approval of eteplirsen in the US.31 However, the amount of new functional dystrophin required to produce a clinically meaningful outcome has yet to be quantified.32 In some rare cases, the absence of dystrophin does not cause severe disease, as would be predicted.33
There are also questions regarding the reliability and consistency of the current methods used to quantify dystrophin production.33,34 Different samples using different techniques can yield varying results that cannot be compared, as they have been tested at different times, with different methods, and at different testing sites. For example, the average dystrophin protein level after 180 weeks of treatment with eteplirsen was increased by only 0.93% ± 84% when tested by Western Blot in Study 202.31,33
All RCTs were double-blinded and placebo-controlled. However, one important limitation is the duration of the trials. In studies of less than 52 weeks, the 6MWT is more likely to detect changes in patients who are in a rapid decline phase of the disease.15 The production of dystrophin may take months, but demonstrating a positive clinical outcome such as a delay to a non-ambulatory state and improved life expectancy may take years to demonstrate.34
Safety
Eteplirsen
In the group that received treatment (n = 8), the US prescribing information indicates that the most frequent adverse events reported in Study 201 (n = 12) were balance disorder (38%; n = 3), vomiting (25%; n = 2), and contact dermatitis (25%; n = 2). No adverse events were reported in the placebo group (0%; n = 0).31
The US monograph further states that in combined clinical studies of patients who received eteplirsen 30 mg/kg (n = 88) for up to 208 weeks, adverse events that reached a frequency ≥ 10% included vomiting, contusion, excoriation, arthralgia, rash, catheter-site pain, and upper respiratory tract infection.31
Transient infusion–related reactions such erythema, facial flushing, and elevated temperature have also been reported.31
Ataluren
Frequent adverse events reported in Study 007 were vomiting (ataluren 40 mg/kg: 56.1%; placebo: 38.6%), diarrhea (ataluren 40 mg/kg: 19.3%; placebo: 24.6%), nausea (ataluren 40 mg/kg: 14.0%; placebo: 12.3%), and upper abdominal pain (ataluren 40 mg/kg: 15.8%; placebo: 15.8%). Serious adverse events in Study 007 were reported in 3.5% of the ataluren 40 mg/kg arm and 5.3% in the placebo arm. None of the participants withdrew from the study due to treatment-related adverse events.26 Exposure to ataluren may cause elevation of liver enzymes, serum lipids, and serum creatinine; these effects were reversible upon discontinuation.35 Concomitant administration with aminoglycosides is contraindicated.15,26
Idebenone
No deaths were reported in the DELOS trial.17 Serious adverse events were reported in 6% of idebenone-treated patients and in 15% of placebo patients. Withdrawals due to adverse events were reported in two idebenone patients (due to sleep apnea syndrome in one patient and diarrhea in another patient) and in one placebo patient (due to supraventricular arrhythmia and respiratory failure with pneumonia). The most frequently reported adverse events included nasopharyngitis (26%) and headache (20%), which were balanced across treatment groups. Diarrhea was more commonly reported in the idebenone group compared with placebo (25% versus 12%, respectively).17
Post-market reports of reduced blood cell counts and increased liver function have been reported (in the indication for Friedreich’s ataxia).36
Administration and Cost
Eteplirsen
In the US, eteplirsen is available in 100 mg/2 mL and 500 mg/10 mL single-dose vials. The recommended dose is 30 mg/kg administered once weekly as a 35-minute to 60-minute intravenous infusion.31 The cost of eteplirsen is estimated to be approximately US$300,000 per year.37
Ataluren
In Europe, ataluren is available in sachets containing oral granules for suspension in strengths of 125 mg, 250 mg, and 1,000 mg. The granules can be taken orally after mixing with either a liquid or semi-solid food such as yogurt. The recommended dosage of 40 mg/kg/day is to be divided into three doses (10 mg/kg in the morning, 10 mg/kg at midday, and 20 mg/kg in the evening).38
The projected benchmark cost in Canada is $264 for a 250 mg sachet and $1,056 for the 1,000 mg sachet.39 Assuming a weight of 25 kg, the cost per year would be $385,440.
Idebenone
Idebenone is administered as two 150 mg film-coated tablets that are given orally three times a day (900 mg/day) with meals.40 The cost of idebenone has yet to be determined.
Concurrent Developments
AONs targeting exon 51 are the most advanced exon-skipping therapy in clinical development; other similar therapies designed to skip mutations on other exons (44, 45, 53, 56) are in the initial stages of clinical trials.7,41,42
The use of adeno-associated virus (AAV) vectors to deliver working copies of the DMD gene to muscles is being investigated; however, early clinical results have been unremarkable and this type of therapy presents some challenges, such as immune reactions to the newly translated protein.3,7
An emerging tool of bacterial origin for correcting mutations is gene editing with clustered regularly interspaced short palindromic repeats. This method has been successfully paired with AAV vectors to treat dystrophic mice with positive initial results.43
CAT-1004, an orally administered small molecule targeted to inhibit activated NF-kappaB, a major inflammatory and muscle degeneration factor implicated in DMD pathophysiology, is being investigated in phase I and phase II studies.44
Vamorolone, an anti-inflammatory and potential alternative to prednisone, is being investigated in phase II trials with the hope that it may have the beneficial effects of prednisone with reduced side effects.45,46
Lastly, inhibition of myostatin, which is a negative inhibitor of muscle mass, is a novel strategy in regenerating muscle in muscular dystrophy patients. ACE-031, a fusion protein designed to bind and disrupt the activity of myostatin, was investigated and showed a trend for improvements in the 6MWT; however, the study was stopped due to adverse events of epistaxis and telangiectasias.47 Other trials investigating different myostatin inhibitors, such as follistatin and PF-06252616 (phase II), are being conducted.6,48
Implementation Issues
Current access to DMD drugs is limited, as patients in Canada are generally restricted to participation in clinical trials. Patient organizations such as TREAT-NMD and Muscular Dystrophy Canada may help eligible patients participate in trials, although many studies are recruiting by invitation only.6
Once these medications receive regulatory approval, a major barrier to the uptake of novel therapies for DMD is likely to be cost. Furthermore, there is the possibility for some of the medications to be used in combination, which will increase the cost further. Criteria will need to be determined to establish which patients are more likely to benefit, and to identify when a drug is no longer beneficial.
Administering medication to children can be challenging. Idebenone and ataluren are orally administered and require multiple daily administrations. The oral route of administration may not be ideal in DMD patients who have trouble swallowing, although ataluren has the advantage of being available as granules for suspension. Eteplirsen needs to be infused intravenously once weekly, which could become a barrier, depending on accessibility and availability of treatment centres.
Acknowledgments
CADTH thanks the external reviewers who kindly provided comments on an earlier draft of this bulletin.
Footnotes
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While CADTH has taken care to ensure that the information prepared by it in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. You are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
References
- 1.
- de los Angeles Beytía M, Vry J, Kirschner J. Drug treatment of Duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol [Internet]. 2012. pp. 4–8. [cited 2016 Dec13] Available from: https://www
.ncbi.nlm .nih.gov/pmc/articles /PMC3440798/pdf/1128-2460-31-4.pdf. [PMC free article: PMC3440798] [PubMed: 22655510] - 2.
- Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014 Jun;24(6):482–91. [PubMed: 24780148]
- 3.
- Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol [Internet] 2015 Dec;100(12):1458–67. [cited 2017 Jan 6] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4973818. [PMC free article: PMC4973818] [PubMed: 26140505] - 4.
- Canadian Neuromuscular Diseases Network, Canadian Neuromuscular Disease Registry. Duchenne muscular dystrophy (DMD) in Canada [Internet]. Toronto: Muscular Dystropy Canada; 2016. [cited 2016 Nov 2]. Available from: http://www
.muscle.ca /wp-content/uploads/2012 /11/CNDR_DMD_in_Canada_Report.pdf. - 5.
- Abdul-Razak H, Malerba A, Dickson G. Advances in gene therapy for muscular dystrophies. F1000Res [Internet]. 2016. [cited 2017 Apr 19] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4991540. [PMC free article: PMC4991540] [PubMed: 27594988] - 6.
- Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat [Internet]. 2016. pp. 1795–807. [cited 2016 Nov 2] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4966503. [PMC free article: PMC4966503] [PubMed: 27524897] - 7.
- Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am J Transl Res [Internet] 2016;8(6):2471–89. [cited 2016 Nov 2] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4931144. [PMC free article: PMC4931144] [PubMed: 27398133] - 8.
- Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51(8):759–64. [PubMed: 25752877]
- 9.
- Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. 2015 Jun 29;87:104–7. [PubMed: 25980936]
- 10.
- Ataluren (Translarna™) [Internet]. PTC Therapeutics Inc.; 2016. [cited 2016 Nov 2]. Available from: http://www
.ptcbio.com /en/pipeline/ataluren-translarna. - 11.
- Summary of cancellation: ataluren (*Translarna). Ottawa: Health Canada; 2016.
- 12.
- FDA rejects BioMarin’s muscle wasting drug; Sarepta drug in focus [Internet]. Sunnyvale (CA): Yahoo Inc.; Jan 14, 2016. [cited 2016 Nov 2]. Available from: http://finance
.yahoo .com/news/fda-rejects-biomarins-dmd-drug-135156448.html. - 13.
- FDA grants accelerated approval to first drug for Duchenne muscular dystrophy [Internet]. Silver Spring (MD): U.S. Food and Drug Administration; Sep 19, 2016. [cited 2016 Nov 2]. (FDA news release). Available from: http://www
.fda.gov/NewsEvents /Newsroom/PressAnnouncements /ucm521263.htm. - 14.
- Bushby K, Finkel R, Wong B, Barohn R, Campbell C, Comi GP, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve [Internet] 2014 Oct;50(4):477–87. [cited 2016 Dec 13] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4241581. [PMC free article: PMC4241581] [PubMed: 25042182] - 15.
- Translarna™ (ataluren) 125, 250, 1000 mg. Dublin (IE): PTC Therapeutics International Ltd.; Dec 16, 2016.
- 16.
- Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013 Nov;74(5):637–47. [PubMed: 23907995]
- 17.
- Buyse GM, Voit T, Schara U, Straathof CS, D’Angelo MG, Bernert G, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet [Internet] 2015 May 2;385(9979):1748–57. [cited 2016 Dec 13] Available from: http://www
.sciencedirect .com/science/article /pii/S0140673615600253. [PubMed: 25907158] - 18.
- Mcdonald CM, Meier T, Voit T, Schara U, Straathof CS, D’Angelo MG, et al. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul Disord [Internet] 2016 Aug;26(8):473–80. [cited 2016 Dec 13] Available from: http://www
.sciencedirect .com/science/article /pii/S0960896616301572. [PubMed: 27238057] - 19.
- Tinsley J, Muntoni F, Spinty S, Roper H, Hughes I, Ricotti V, et al. Utrophin modulators to treat duchenne muscular dystrophy (DMD): Phase 1B clinical trial results of SMT c1100 [abstract]. Neurology [Internet]; Presented at 67th American Academy of Neurology Annual Meeting, AAN 2015; 2015 Apr 18–25; Washington, DC. 2015. p. 84. [cited 2016 Dec 14] Available from: http://www
.neurology .org/content/84/14_Supplement/P2.006. - 20.
- Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2016 Oct;26(10):643–9. [PubMed: 27566866]
- 21.
- Douglas AGL, Wood MJA. Splicing therapy for neuromuscular disease. Molecular and Cellular Neuroscience [Internet]. 2013. pp. 169–85. [cited 2016 Dec 13] Available from: https://www
.ncbi.nlm .nih.gov/pmc/articles/PMC3793868/ [PMC free article: PMC3793868] [PubMed: 23631896] - 22.
- Darras BT. Post TW, editor. Waltham (MA): UpToDate; Oct 27, 2016. Treatment of Duchenne and Becker muscular dystrophy. [Internet] [cited 2016 Jan 6] Available from: www
.uptodate.com Subscription required. - 23.
- Darras BT. Post TW, editor. UpToDate [Internet]. Waltham (MA): UpToDate; Jan 21, 2016. Clinical features and diagnosis of Duchenne and Becker muscular dystrophy. [cited 2016 Jan 6]. Available from: www
.uptodate.com Subscription required. - 24.
- Mcdonald CM, Henricson EK, Abresch RT, Florence J, Eagle M, Gappmaier E, et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve [Internet] 2013 Sep;48(3):357–68. [cited 2016 Dec 13] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC3826053. [PMC free article: PMC3826053] [PubMed: 23674289] - 25.
- Dubow J, Cunniff T, Wanaski S, Meyer J. Effect of deflazacort and prednisone versus placebo on pulmonary function in boys with duchenne muscular dystrophy who have lost ambulation [abstract]. Ann Neurol [Internet]; Presented at 45th Annual Meeting of the Child Neurology Society; 2016 Oct 26–29; Vancouver, BC. 2016. pp. S370–1. [cited 2016 Dec 14] Available from: http:
//onlinelibrary .wiley.com/doi/10.1002/ana.24756/epdf. - 26.
- Haas M, Vlcek V, Balabanov P, Salmonson T, Bakchine S, Markey G, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord. 2015 Jan;25(1):5–13. [PubMed: 25497400]
- 27.
- Goemans N, Campbell C, Mcdonald CM, Voit T, Luo X, Elfring G, et al. ACTDMD (Ataluren Confirmatory Trial in Duchenne Muscular Dystrophy): Effect of Ataluren on timed function tests (TFT) in nonsense mutation (nm) DMD [abstract]. Can J Neurol Sci [Internet]; Presented at 51st Annual Congress of the Canadian Neurological Sciences Federation; 2016 Jun 21–24; Quebec, QC. 2016. p. S15. [cited 2016 Dec 14] Available from: https://www
.cambridge .org/core/services/aop-cambridge-core /content /view/28EEC950FEA037E7175DA7024585D163 /S0317167116000810a .pdf/div-class-title-d-08-act-dmd-ataluren-confirmatory-trial-in-duchenne-muscular-dystrophy-effect-of-ataluren-on-timed-function-tests-tft-in-nonsense-mutation-nm-dmd-div.pdf. - 28.
- PTC Therapeutics. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Mar 26, 2013. Phase 3 Study of Ataluren in Patients With Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD). [cited 2017 Jan 11, updated 2 Aug 2016] 2000-. Available from: https://clinicaltrials.gov/ct2/show/NCT01826487?term=NCT01826487&rank=1 Identifier: NCT01826487.
- 29.
- Sarepta Therapeutics. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Aug 8, 2011. Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients. [cited 2017 Jan 11, updated 12 Oct 2015] 2000-. Available from: https://clinicaltrials.gov/ct2/show/study/NCT01396239?term=eteplirsen&rank=3 Identifier: NCT01396239.
- 30.
- Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol [Internet] 2016 Feb;79(2):257–71. [cited 2016 Dec 13] Available from: https://www
.ncbi.nlm .nih.gov/pmc/articles/PMC5064753/ [PMC free article: PMC5064753] [PubMed: 26573217] - 31.
- EXONDYS 51 (eteplirsen) injection, for intravenous use [label on the Internet]. Silver Spring (MD): U.S. Food and Drug Administration; Sep, 2016. [cited 2016 Jun 20]. Available from: http://www
.accessdata .fda.gov/drugsatfda_docs /label/2016/206488lbl.pdf. - 32.
- Wood MJ. To skip or not to skip: that is the question for duchenne muscular dystrophy. Mol Ther [Internet] 2013 Dec;21(12):2131–2. [cited 2017 Jan 6] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC3863800. [PMC free article: PMC3863800] [PubMed: 24317541] - 33.
- Dunn B. Eteplirsen for treatment of Duchenne muscular dystrophy in patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping therapy [slide deck on the Internet]. Silver Spring (MD): U.S. Food and Drug Administration; Apr 25, 2016. [cited 2017 Jan 11]. Available from: http://www
.fda.gov/downloads /AdvisoryCommittees /CommitteesMeetingMaterials /Drugs /PeripheralandCentralNervousSystemDrugsAdvisoryCommittee /UCM500821.pdf. - 34.
- Merlini L, Sabatelli P. Improving clinical trial design for Duchenne muscular dystrophy. BMC Neurol [Internet]. Aug 26, 2015. p. 153. [cited 2017 Jan 6] Available from: http://www
.ncbi.nlm.nih .gov/pmc/articles/PMC4549867. [PMC free article: PMC4549867] [PubMed: 26306629] - 35.
- Campbell C, Shieh P, Sejersen T, Luo X, Elfring G, Kroger H, et al. Safety and tolerability of ataluren in a phase 3 study of patients with nonsense mutation duchenne muscular dystrophy [abstract]. Neurology [Internet]; Presented at 68th American Academy of Neurology Annual Meeting, AAN 2016; 2016 Apr 15–21; Vancouver, BC. 2016. p. 16. [cited 2016 Dec 14] Available from: http://www
.neurology .org/content/86/16_Supplement/P3.164. - 36.
- Summary basis of decision (SBD) for PrCATENA®. Ottawa: Health Canada; Mar 6, 2009.
- 37.
- BioMarin announces withdrawal of market authorization application for Kyndrisa™ (drisapersen) in Europe [Internet]. San Rafael (CA): BioMarin Pharmaceutical Inc.; May 31, 2016. [cited 2016 Nov 2]. Available from: https:
//globenewswire .com/news-release/2016 /05/31/844933/0/en /BioMarin-Announces-Withdrawal-of-Market-Authorization-Application-for-Kyndrisa-drisapersen-in-Europe.html. - 38.
- Translarna (ataluren) [Internet]. London: European Medicines Agency; Aug, 2014. [cited 2017 Jan 11]. (EPAR summary for the public). Available from: http://www
.ema.europa .eu/docs/en_GB/document_library /EPAR_-_Summary _for_the_public /human/002720/WC500171815.pdf. - 39.
- Husain M. Letter to: Paula Murray [Internet]. Dublin (IE): PTC Therapeutics International Ltd.; Dec 22, 2016. [cited 2016 Sep 29]. Available from: https://www
.ccmg-ccgm .org/images/pdf/DTC-letter-Final-April-15-2015_V6.pdf Subject: CADTH project EH0049-000 emerging drugs for Duchenne muscular dystrophy. - 40.
- Buyse GM, Goemans N, van den Hauwe M, Meier T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr Pulmonol. 2013 Sep;48(9 suppl 2):912–20. [PubMed: 23129412]
- 41.
- Saito T, Nagata T, Masuda S, Suzuki M, Nakamura H, Komaki H, et al. First-in-human study of NS-065/NCNP-01; the morpholino based antisense oligonucleotide for exon 53 skipping in duchenne muscular dystrophy [abstract]. Molecular Therapy; Presented at 18th Annual Meeting of the American Society of Gene and Cell Therapy, ASGCT 2015; 2015 May 13–16; New Orleans, LA. 2015. pp. S55–6.
- 42.
- A phase I/IIa clinical trial in Duchenne muscular dystrophy using systemically delivered morpholino antisense oligomer to skip exon 53 (SKIP-NMD). Hum Gene Ther Clin Dev. 2015 Jun;26(2 suppl 2):92–5. [PubMed: 26086759]
- 43.
- Nelson C, Gemberling M, Hakim CH, Ousterout DG, Thakore PI, Castellanos RM, et al. Local and systemic gene editing in a mouse model of duchenne muscular dystrophy [abstract]. Molecular Therapy; Presented at 19th Annual Meeting of the American Society of Gene and Cell Therapy, ASGCT 2016; 2016 May 4–7; Washington, DC. 2016. p. S191.
- 44.
- Donovan J, Sweeney H, Vandenborne K, Russman B, Jirousek M, Finkel R. CAT-1004, an oral agent targeting NF-kappaB in development for treatment of Duchenne muscular dystrophy: Phase 1/2 study design [abstract]. Neuromuscul Disord; Presented at 20th International Congress of The World Muscle Society; 2015 Oct 1–4; Brighton, United Kingdom. 2015. p. S262.
- 45.
- Vamorolone - a potential steroid alternative customized for Duchenne [Internet]. DuchenneConnect; Jul 29, 2016. [place unknown] [cited 2017 Jan 11]. Available from: https://www
.duchenneconnect .org/clinical-trials /research-faqs /685-vamorolone-a-potential-steroid-alternative-customized-for-duchenne.html. - 46.
- ReveraGen BioPharma, Inc. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Apr 28, 2016. 2000. A study to assess vamorolone in boys with Duchenne muscular dystrophy (DMD). [cited 2017 Jan 11; 20 Dec 2016] Available from: https://clinicaltrials.gov/ct2/show/NCT02760264?term=canada+and+neuromuscular&recr=Open&cntry1=NA%3ACA&rank=83%20Identifier:%20NCT02760264.
- 47.
- Campbell C, McMillan HJ, Mah JK, Tarnopolsky M, Selby K, McClure T, et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2016 Jul 27;(suppl 2) [PubMed: 27462804]
- 48.
- Pfizer. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Nov 4, 2014. 2000. A Phase 2 Study to Evaluate the Safety, Efficacy, Pharmacokinetics and Pharmacodynamics of PF-06252616 in Duchenne Muscular Dystrophy. [cited 2017 Jan 11; 6 Jan 2017] Available from: https://clinicaltrials.gov/ct2/show/NCT02310763?term=NCT02310763&rank=1 Identifier: NCT02310763.
- 49.
- Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules. 2015 Oct 7;20(10):18168–84. [PMC free article: PMC6332113] [PubMed: 26457695]
- 50.
- Govoni A, Magri F, Brajkovic S, Zanetta C, Faravelli I, Corti S, et al. Ongoing therapeutic trials and outcome measures for Duchenne muscular dystrophy. Cellular and Molecular Life Sciences. 2013;70(23):4585–602. [PubMed: 23775131]
- 51.
- What is exon skipping and how does it work? [Internet]. London: Muscular Dystrophy UK; 2016. [cited 2016 Nov 2]. Available from: http://www
.musculardystrophyuk .org/progress-in-research /background-information /what-is-exon-skipping-and-how-does-it-work/ - 52.
- Lewis R. Exon skipping: borrowing from nature to treat rare genetic diseases. San Francisco: PLoS Blogs; Feb 20, 2014. DNA science blog: genetics in context [blog on the Internet] [cited 2016 Nov 2]. Available from: http://blogs
.plos.org /dnascience/2014/02 /20/exon-skipping-borrowing-nature-treat-rare-genetic-diseases/ - 53.
- Quinlivan R, Luo X, Elfring G, Kroger H, Riebling P, Ong T, et al. Ataluren confirmatory trial in dmd: Effect of ataluren on activities of daily living in nonsense mutation duchenne muscular dystrophy [abstract]. Neurology; Presented at 68th American Academy of Neurology Annual Meeting, AAN 2016; 2016 Apr 15–21; Vancouver, BC. 2016.
- 54.
- Tinsley J, Robinson N, Davies KE. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J Clin Pharmacol [Internet] 2015 Jun;55(6):698–707. [cited 2016 Dec 13] Available from: https://www
.ncbi.nlm .nih.gov/pmc/articles/PMC5024067/ [PMC free article: PMC5024067] [PubMed: 25651188] - 55.
- Duchenne muscular dystrophy [Internet]. Abingdon (GB): Summit Therapeutics; 2016. [cited 2016 Nov 2]. Available from: http://www
.summitplc .com/programmes/duchenne-muscular-dystrophy/ - 56.
- RAXONE® - Phase 3 study assessing the efficacy, safety and tolerability of idebenone in patients with Duchenne muscular dystrophy receiving glucocorticoid steroids (SIDEROS) [Internet]. place unknown: DuchenneConnect; May 9, 2016. [cited 2016 Nov 2]. Available from: https://www
.duchenneconnect .org/clinical-trials /research-faqs /652-catenar-phase-3-study-of-idebenone-in-duchenne-muscular-dystrophy-delos .html. - 57.
- GIVINOSTAT (ITF2357) - A histone deacetylase (HDAC) inhibitor for the treatment of Duchenne [Internet]. DuchenneConnect; Apr 22, 2016. [place unknown] [cited 2016 Nov 2]. Available from: https://www
.duchenneconnect .org/clinical-trials /research-faqs /991-givinostat-itf2357-a-histone-deacetylase-hdac-inhibitor-for-the-treatment-of-duchenne.html. - 58.
- Sarepta Therapeutics. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Sep 25, 2014. Confirmatory study of eteplirsen in DMD patients (PROMOVI). [cited 2017 Jan 11, updated 27 Dec 2016] 2000-. Available from: https://clinicaltrials.gov/ct2/show/NCT02255552?term=eteplirsen&rank Identifier: NCT02255552.
- 59.
- Santhera Pharmaceuticals. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Jun 17, 2016. A Phase III double-blind study with idebenone in patients with Duchenne muscular dystrophy (DMD) taking glucocorticoid steroids (SIDEROS). [cited 2016 Nov 2; last updated: 2016 Oct 17] 2000-. Available from: https://www
.clinicaltrials .gov/ct2/show?term =Idebenone&cond= %22Duchenne+Muscular+Dystrophy %22&rank=4 Identifier: NCT02814019. - 60.
- Italfarmaco. ClinicalTrials.gov [Internet]. Bethesda (MD): U.S. National Library of Medicine; Sep 25, 2014. Clinical study to evaluate the efficacy and safety of givinostat in ambulant patients with Duchenne muscular dystrophy. [cited 2017 Jan 11; updated 28 Jul 2016] 2000-. Available from: https://clinicaltrials.gov/ct2/show/NCT02851797?term=givinostat&cond=duchenne&rank=1 Identifier: NCT02851797.
Appendix 1. Mechanism of Action
The Duchenne muscular dystrophy (DMD) gene is expressed mainly in skeletal and cardiac muscle and is the largest known human gene.49 It contains 79 exons and is subject to complex and numerous mutations due to its large size. While the majority of DMD cases are maternally inherited, one-third result from spontaneous mutations that vary in severity.6,7
The majority of DMD mutations are large mutations that involve one or more exons. Approximately 86% of these are deletions, and 14% are duplications.5,7 The remainder are small mutations, including insertions, nonsense mutations that cause premature cessation of the protein formation, and splicing mutations.6,7,49
Exon Skipping
The method of exon skipping utilizes antisense oligonucleotides (AONs) sequences to skip the exons with mutations, with the aim of correcting specific gene mutations.49,50 This results in partial production of a truncated dystrophin protein and, consequently, partially preserves dystrophin function.50–52 Exon skipping works in cases of deletions, but is less amenable to correcting duplications because the AONs cannot differentiate between the original and duplicated exons.7 The most frequent mutations on the DMD gene occur between exons 45 and 55, and therapy aimed at skipping exon 51 has been of particular interest, as it accounts for 13% of DMD mutations.16 Figure 1 depicts the use of an AON to target a specific exon and restore the reading frame in order to enable the production of a partially truncated dystrophin.
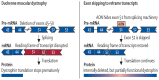
Figure 1
Exon Skipping by Antisense Oligonucleotides to Restore Reading Frame AON = antisense oligonucleotide; mRNA= messenger ribonucleic acid.
Eteplirsen is a third-generation synthetic AON that is designed to induce exon 51 skipping. There are several other exon-skipping therapies in clinical trials that target different exons: exon 44 (PRO044); exon 45 (DS-5141b, PRO045, SRP4045); and exon 53 (SRP4053, PRO053, NS-065/NCNP-01).6,7
Of note, drisapersen (Kyndrisa by Biomarin Pharmaceutical), a second-generation AON that induces exon 51 skipping, was rejected by the FDA in January 2016 after it concluded that a standard of evidence of effectiveness had not been met.12 On May 31, 2016, BioMarin announced that it was discontinuing clinical and regulatory developments of Kyndrisa, as well as three other products: BMN 044, BMN 045, and BMN 053, which were in phase II studies for DMD.37
Stop-Codon Read-Through Nonsense Suppression Therapy
This treatment is applicable to approximately 10% to 15% of males with DMD who have a point mutation involving a premature stop codon.1,49 This particular mutation causes the translation of the dystrophin protein to be stopped prematurely, and ultimately produces a shortened and dysfunctional dystrophin protein.
Ataluren was developed to target these mutations by enabling read-through of the premature stop codon and enabling translation and production of a modified dystrophin protein.6,49,53 The resulting increase in dystrophin production would be expected to stabilize or slow disease progression. Ataluren’s mechanism selects only premature stop codons; it does not interfere with normal stop codons.15
Utrophin Upregulation
Utrophin has structural and protein-binding properties similar to dystrophin and shares 80% of its sequence.1,49,54 It is produced during the early stages of muscle-fibre development and is switched off in maturing muscle fibres, at which point dystrophin is produced. When muscle fibre is damaged, utrophin expression is switched on again and production is increased to re-establish the continuity of myotubes and promote the repair mechanism.
Ezutromid is an utrophin modulator that aims to replace lost dystrophin with utrophin.54 Utrophin modulators have the advantage of being potentially applicable to all DMD patients, irrespective of the underlying genetic defect.55 Ezutromid is at the phase IIa development stage.
Adenosine Triphosphate Modulation
Idebenone is a short-chain benzoquinone that increases the energy output of cell mitochondria. It carries and drops off electrons within the mitochondria numerous times, thereby providing additional electrons to the mitochondria. It is also an antioxidant and can neutralize destructive free radicals in cells. All of these activities help dystrophic muscle cells maintain their cellular energy supply, and they protect the cells from oxidative stress.56
Idebenone is an adenosine triphosphate production modulator and is aimed at DMD patients when they become non-ambulatory and their respiratory function begins to significantly decline (ages 10 to 18).40 It is currently being tested in DMD patients for slowing the loss of pulmonary function (Appendix 2).
Histone Deacetylase Inhibitor Activity
Histone deacetylase inhibitor activity is upregulated in dystrophic muscles and contributes to the impairment of muscle regeneration. Givinostat is a histone deacetylase inhibitor currently being studied for the treatment of DMD (Appendix 2). Givinostat promotes the transcription of a number of factors that are important in muscle regeneration, such as follistatin. The expected effects are an increase in muscle mass and the prevention of muscle degeneration. Givinostat also has potent anti-inflammatory effects; these activities may help to rebalance the repair process in muscle with increased muscle regeneration and reduced fatty infiltration and fibrosis.57
A phase I single-arm trial showed that givinostat did not improve outcomes such as the six-minute walk test and NSAA.20 A phase III trial is being planned (Appendix 2).
Appendix 2. Characteristics and Results of Phase IIb and Phase III Clinical Trials
Table
placebo = −4.0% (2.92) eteplirsen 30 mg/kg = 22.9% (2.90)
Appendix 3. Ongoing Phase III Clinical Trials for Duchenne Muscular Dystrophy
Publication Details
Author Information and Affiliations
Authors
Fady Shawi, Christine Perras, and Melissa Severn.Publication History
Published online: June 1, 2017.
Copyright
Except where otherwise noted, this work is distributed under the terms of a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence (CC BY-NC-ND), a copy of which is available at https://creativecommons.org/licenses/by-nc-nd/4.0/
Publisher
Canadian Agency for Drugs and Technologies in Health, Ottawa (ON)
NLM Citation
Shawi F, Perras C, Severn M. Emerging Drugs for Duchenne Muscular Dystrophy. 2017 Jun 1. In: CADTH Issues in Emerging Health Technologies. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2016-2021. 161.
