

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

INTRODUCTION
Thyrotoxicosis is defined as the clinical syndrome of hypermetabolism resulting from increased free thyroxine (T4) and/or free triiodothyronine (T3) serum levels (1). The term thyrotoxicosis is not synonymous with hyperthyroidism, the elevation in thyroid hormone levels caused by an increase in their biosynthesis and secretion by the thyroid gland (Table 1) (2). For example, thyrotoxicosis can result from the destruction of thyroid follicles and thyrocytes in the various forms of thyroiditis, or it can be caused by an excessive intake of exogenous thyroid hormone. It should also be noted that the elevation of free thyroid hormone levels does not always result in thyrotoxicosis in all tissues. In the syndrome of Resistance to Thyroid Hormone (RTH), dominant negative mutations in the thyroid hormone receptor bets (TRbeta) result in decreased thyroid hormone action in tissues where TRbeta is the predominant receptor, for example in the liver and the pituitary, whereas other tissues such as the heart, which express mainly TRalpha, show signs of increased thyroid hormone action (See Chapter 16 D). The most common form of thyrotoxicosis is Graves' disease, which is discussed in Chapter 10. This chapter reviews other etiologies of thyrotoxicosis (Table 1). The determination of the etiology of thyrotoxicosis is of importance in order to establish a rational therapy.
Table 1Etiologies of thyrotoxicosis
| A) Thyrotoxicosis caused by hyperthyroidism | |
| Entity | Pathogenesis |
| Graves' disease | TSH receptor-stimulating antibodies |
| Toxic adenoma | Somatic gain-of-function mutations in the TSH receptor or Gs |
| Toxic multinodular goiter | Somatic gain-of-function mutations in the TSH receptor or Gs |
| Hyperthyroid thyroid carcinoma | Somatic gain-of-function mutations in the TSH receptor |
| Familial non-autoimmune hyperthyroidism | Germline gain-of-function mutations in the TSH receptor |
| Sporadic non-autoimmune hyperthyroidism | Germline gain-of-function mutations in the TSH receptor |
| TSH secreting pituitary adenoma | Increased stimulation by inappropriate TSH secretion |
| hCG-induced gestational hyperthyroidism | Increased stimulation of the TSH receptor by hCG |
| Familial hypersensitivity to hCG | TSH receptor mutation with increased sensitivity to hCG |
| Trophoblast tumors (hydatiform mole, choriocarcinoma) | Increased stimulation of the TSH receptor by hCG |
| Struma ovarii | Autonomous function of thyroid tissue in ovarian teratoma |
| Iodine-induced hyperthyroidism | Increased synthesis of thyroid hormone in autonomously functioning thyroid tissue after exposure to excessive amounts of iodide |
| B) Thyrotoxicosis without hyperthyroidism | |
|---|---|
| Subacute thyroiditis | Release of stored thyroid hormone |
| Silent thyroiditis | Release of stored thyroid hormone |
| Drug-induced thyroiditis | Release of stored thyroid hormone |
| Exogenous thyroid hormone (iatrogenic, thyrotoxicosis factitia) | Thyroid hormone |
TSH: Thyroid-stimulating hormone. Gs: stimulatory G protein subunit. hCG = human chorionic gonadotropin.
TOXIC ADENOMA
Definition and Epidemiology
A toxic adenoma is a monoclonal, autonomously functioning thyroid nodule (AFTN) that produces supraphysiological amounts of T4 and/or T3 resulting in suppression of serum TSH. The function of the surrounding normal thyroid tissue is often, but not always, suppressed. Approximately 1 in 10 to 20 solitary nodules present with hyperthyroidism. The prevalence of hyperthyroidism appears to be more common in Europe than in the USA, and it is more common in women than in men (3, 4). In a series of 349 patients with AFTNs, 287 were nontoxic and 62 were toxic (3). Toxic lesions were seen in 56.5% of patients over 60 years, but in only 12.5% of the younger patients. The female to male ratio was 14.9:1 for nontoxic AFTNs and 5.9:1 for toxic AFTN patients. T3 thyrotoxicosis was observed in 46% of the patients with hyperthyroidism. All but 4 of the toxic AFTN measured 3 cm in diameter or were larger. AFTNs 3 cm or larger were more than twice as common in patients 40 years or older than in younger patients. Of 159 untreated nontoxic AFTN patients, 14 became toxic within 1 to 6 years (3). In a study from Switzerland on 306 patients with toxic adenomas, the female to male ratio was 5: 1 (5). The frequency of toxic adenomas in patients referred for thyrotoxicosis varies considerably in different geographical areas and appears to be more common in countries with insufficient nutritional iodide intake; reported percentages vary between 1.5 and 44.5% (6). In a prospective European multicenter study (17 centers in 6 countries) 924 untreated hyperthyroid patients were investigated (2). 9.2% of the patients had an autonomous adenoma and 59.6% had Graves' disease. Among the 31.2% with unclassified hyperthyroidism, a majority probably also had Graves' disease. Autonomous adenomas were more frequent in iodine-deficient areas (10.1%) than in iodine-sufficient areas (3.2%) (2). In a Swedish study, the mean annual incidence of toxic adenomas (4.8 per 100,000) did not differ between 1988 and 1990 and between 1970 and 1974 (7).
Clinical presentation
Patients with toxic adenomas present with signs and symptoms of thyrotoxicosis and/or a thyroid nodule. The signs and symptoms of thyrotoxicosis do not differ from other etiologies. Features suggestive for Graves' disease such as endocrine ophthalmopathy, (pretibial) myxedema and acropachy are missing. The onset of thyrotoxicosis is often insidious and more common in older patients, who typically have larger adenomas. However, a toxic adenoma has even been documented as a cause of neonatal hyperthyroidism (8). Mechanical symptoms such as dysphagia or hoarseness are uncommon. Autonomously functioning nodules may remain stable in size, grow, degenerate or become gradually toxic. In one series, 10% of patients followed for 6 years became thyrotoxic (3). Thyrotoxicosis may develop independent of age, but is much more common in nodules ov er 3 cm in diameter (up to 20%). By sonography, the critical volume at which hyperthyroidism occurs is about 16 ml (9). Changes in nodule size were followed in 159 patients during a period of 1 to 15 years (3). An increase in size was seen in only 10%, 4% of nodules decreased, and a loss of function due to degenerative changes was observed in 4 nodules. Eight percent developed overt thyrotoxicosis during a follow-up of 3 to 5 years, and 3% developed subclinical hyperthyroidism (3).
Diagnosis

The measurement of serum TSH with a sensitive third-generation assay represents the best biochemical marker to establish the diagnosis of thyrotoxicosis because TSH and FT4 have an inverse log-linear relationship and a small decrease or increase in FT4 is thus associated with an exponential change in TSH levels (10). If the TSH is suppressed, measurement of serum (free) T4 and T3 permit to ascertain the severity of the thyroid overactivity. A thyroid scan can be performed with 123iodine, 131iodine, or 99technetium-labeled pertechnetate (11). Iodine isotopes, which are not only trapped but also organified in the thyroid, are preferred because 3-8% of nodules that appear functioning on pertechnetate scanning are nonfunctioning on radioiodine scanning. A scan will show uptake of the isotope that is either limited to the nodule, or preferential uptake in the adenoma compared to the surrounding tissue (Figure 1). Scintigraphically, an AFTN may be warm (uptake similar to surrounding tissue), hot (uptake increased without suppression of surrounding tissue), or toxic (uptake increased and suppression of the surrounding tissue). A toxic nodule is associated with overt or subclinical hyperthyroidism. A warm nodule may develop into a hot nodule and ultimately into a toxic adenoma. Toxic adenomas are usually larger in size and often more than 3 cm in size (3, 12). In the case of incomplete suppression of the surrounding tissue, autonomous function of the nodule can be established by a suppression test. After the administration of thyroid hormone (e.g. 75 �g of levothyroxine for 2 weeks, followed by 150 �g for 2 weeks), a repeat thyroid scan would fail to show remaining uptake in the non-autonomous tissue because of the suppression of serum TSH, thereby unmasking the autonomy of the nodule (13). However, this procedure has no practical consequences and is therefore unnecessary in clinical practice. Ultrasound will confirm the presence of a solitary nodule and may show a small contralateral thyroid lobe. There is no indication to perform fine needle aspiration in patients with toxic adenomas because the risk of a thyroid carcinoma is extremely low and cytological evaluation will not permit distinguishing between a follicular adenoma and a follicular carcinoma (14, 15).
Treatment
In patients with overt thyrotoxicosis, definitive forms of treatment include surgical excision of the nodule, treatment with radioactive iodine, or percutaneous ethanol injection (11, 16). Treatment with antithyroid drugs is used infrequently as it requires long-term therapy and a relapse will almost invariably occur after discontinuation of the medication. Surgical excision permits to achieve a rapid and permanent control of hyperthyroidism with a very low operative complication rate. The disadvantage of a surgical approach includes the risks of general anesthesia and the potential complications of thyroid surgery. Usually the patient is treated preoperatively with antithyroid drugs and beta-blockers. The incidence of hypothyroidism after operation is low, but may occur. In a series of 60 patients operated for AFTNs, 6.6% became hypothyroid after operation (17). Two of these patients had previously received therapeutic doses of 131iodine or long-term treatment with antithyroid drugs. In a series of 35 patients with a solitary toxic adenoma, lobectomy resulted in 30 euthyroid and 5 hypothyroid outcomes, although hypothyroidism was only temporary in 3 patients (4). It remains unclear why some of these patients remained permanently hypothyroid after lobectomy; information about the presence of autoantibodies and the morphology of the contralateral lobe is not provided in this study. Generally, it is believed that long-term suppression of the thyroid gland does not lead to permanent inactivation after suppression is relieved. Administration of 131iodine is a widely used therapeutic modality for patients with toxic adenomas. The main disadvantage consists in the possibility of permanent hypothyroidism in a subset of patients. In a study by Goldstein et al., 23 patients were followed for 4 to 16.5 years and 8/23 (35%) developed hypothyroidism (18). The incidence of hypothyroidism was not related to nodule size, the level of thyroid function, or the administered dose of 131iodine. In a similar study by Mariotti et al. on 126 patients, 5/126 (4%) developed overt hypothyroidism 1 to 10 years after 131iodine therapy (19). There was no relationship between the development of hypothyroidism, nodule size or the administered dose of 131iodine (19). Hypothyroidism occurred in 9.7% of patients with an euthyroid hot nodule treated with 131iodine, and in only 1.5% of patients with a toxic adenoma. When antithyroglobulin and/or antithyroid microsomal antibodies were present, the prevalence of hypothyroidism after 10 years was 18% versus 1.4% in antibody-negative patients. In two studies, evaluating 48 and 45 patients 6 months after radioiodine therapy, hypothyroidism could not be documented in any of the patients (20, 21). In a more recent study by Bolusani et al. on 105 patients with solitary autonomous nodules, the cumulative incidence of hypothyroidism was 11% at 1 year, 33% at 5 years, and 49% at 10 years (22). The development of hypothyroidism was not associated with age, sex, radioiodine dose, radioiodine uptake, or degree of suppression of extranodal tissue on scintiscans. The predictors of occurrence of hypothyroidism were pretreatment with antithyroid medications and a positive thyroid antibody status. Antibody-positive patients showed an earlier progression towards hypothyroidism than did antibody-negative patients (22). In aggregate, these results suggest that longer follow-up periods may uncover hypothyroidism more frequently and that the development of hypothyroidism may often be related to the presence of thyroid autoantibodies, but less to the administered dose of 131iodine and nodule size. In patients treated with antithyroid drugs prior to radioiodine therapy, the increase in TSH may reactivate suppressed thyroid tissue and iodide uptake resulting in damage by 131iodine. Some clinicians administer levothyroxine for two weeks prior to therapy in order to assure that the tissue surrounding the toxic adenoma is suppressed. In some instances, high doses of 131iodine in the nodule may provide enough radiation to the surrounding tissue that its function is seriously damaged. It is noteworthy that therapy with 131iodine may trigger the development of humoral thyroid autoantibodies (23). For example, about 5% of patients treated with 131iodine for toxic or euthyroid multinodular goiter develop stimulating TSH receptor antibodies and Graves' disease (24). Hence, hypothyroidism may, in part, result from the development of humoral autoantibodies in patients with toxic adenomas treated with 131iodine. An alternative to surgery and 131iodine therapy for toxic adenomas consists in the use of percutaneous ethanol injection into the nodule under ultrasound guidance (11). The injection results in necrosis and thrombosis of small vessels. Side effects include local pain and, in rare cases, recurrent nerve damage. In studies evaluating the outcomes at 12 or 30 months, about 85% of patients were euthyroid (25, 26). Results of ethanol injection in relatively large AFTNs (diameter 3 to 4 cm) are also favorable, particularly in patients with subclinical hyperthyroidism (27, 28). Surpisingly, previous ethanol injection did not hinder histological assessment in 13 patients who ultimately underwent surgical excision of their nodule (29). Percutaneous laser thermal ablation (LTA) is a more recently introduced technique for the debulking of thyroid nodules and has also been used for the debulking of anaplastic thyroid cancer (30). In hyperfunctioning nodules, LTA induced a nearly 50% volume reduction with a variable frequency of normalization of thyroid-stimulating hormone levels (31, 32). Most patients become and remain euthyroid after treatment. However, serum TSH measurement at yearly intervals is necessary in order to detect those patients, especially with circulating thyroid autoantibodies, who will eventually develop hypothyroidism.
Pathogenesis
Chronic stimulation of the cAMP cascade results in enhanced proliferation and function of thyrocytes (33). Hence, any molecular alteration leading to constitutive activation of the cAMP pathway in a thyroid follicular cell is expected to result in clonal autonomous growth and function, and ultimately in a toxic adenoma (33). In line with this concept, somatic mutations were first discovered in the GNAS1 gene encoding the stimulatory Gs alpha subunit in toxic adenomas (34-36). Stimulatory Gs alph amutations impair the hydrolysis of guanine triphosphate (GTP) to guanine diphosphate (GDP), resulting in persistent activation of adenylyl cyclase. Stimulatory Gs alpha mutations are also found in 35-40 percent of somatotroph tumors in acromegalic patients (37), and mosaicism for Gs alpha mutations with onset during blastocyst development causes the McCune Albright syndrome (38). The observation that site-directed mutagenesis of a residue in the third intracellular loop of the alpha 1b-adrenergic receptor can lead to constitutive activation of this G protein-coupled receptor (GPCR) in the absence of ligand led to the search and detection of naturally occurring activating mutations in numerous GPCRs (39). Somatic mutations in the TSH receptor were first discovered in toxic adenomas (40). The initially characterized mutations were clustered in the third intracellular loop and the sixth transmembrane domain of the receptor, but a wide variety of activating somatic mutations have been found in subsequent studies (see Chapter 16 A) (41-44). Mutations conferring constitutive activity occur in the entire transmembrane domain, as well as in the carboxy-terminal region of the extracellular domain. All mutations increase basal cAMP levels, but only a few amino acid substitutions activate the phospholipase C (PLC) cascade in a constitutive manner. Inositoltriphosphate (IP3) accumulation in response to TSH is usually retained. The reported prevalence of TSH receptor mutations in toxic adenomas varies widely, but is as high as 80% (43, 45). For example, in a study on 33 toxic adenomas from 31 patients from Belgium, 27/33 of adenomas were positive for a somatic mutation in the TSH receptor (46). In contrast, in a Japanese study that analyzed the part of the gene encoding the third cytoplasmic loop and the sixth transmembrane segment, only 1/38 toxic adenomas harbored a functionally silent mutation (45). Differences in sampling technique and methodological approach, as well as variations in iodine intake, may contribute to the reported differences (47). It is now well established that somatic, constitutively activating TSH receptor mutations play a predominant role in the pathogenesis of AFTNs, while Gs aalpha mutations are less common (48). It is likely that other somatic mutations are involved in the pathogenesis of the monoclonal toxic adenomas that are negative for mutations in the TSH receptor and Gs alpha(49). Functionally, some of the mutations may alter the positions of the transmembrane helices, thereby mimicking the conformational changes induced by binding of ligand. Alternatively, some mutations may alter the structure of domains that inhibit coupling of the receptor to G proteins in the absence of TSH (50, 51). Activating mutations in the extracellular domain appear to result in a relief of a negative constraint present in the unliganded carboxy-terminal part of the extracellular domain (8, 43, 52-54). It has been suggested that iodine deficiency may be a predisposing factor for the development of AFTNs (55). Based on the fact that autonomous (multi)nodular goiters develop also in iodine-sufficient regions and that there is often a hereditary predisposition, others propose that hereditary and acquired heterogeneity among the thyrocytes play a fundamental role in the pathogenesis of AFTNs and that iodine deficiency only serves as a modulating factor (56).
Pathology
On macroscopic examination, a solitary toxic nodule is surrounded by normal thyroid tissue that is functionally suppressed. Toxic adenomas are histologically classified as encapsulated follicular neoplasms or adenomatous nodules without a capsule (57). Hemorrhage, calcifications and cystic degeneration are commonly present. In a study on 51 solitary adenomas, functional and pathologic characteristics were determined and compared to normal surrounding tissue (58). The adenomas displayed a higher number of cycling cells in the periphery of the adenomas, a high level of iodide trapping because of a high level of sodium/iodide symporter (NIS) gene expression, a high thyroperoxidase (TPO) mRNA and protein content, and low H2O2 generation. The adenomas secreted higher amounts of thyroid hormone than the quiescent tissue (58). The proliferation index was determined in 20 toxic adenomas using labeling with proliferating cell nuclear antigen (PCNA) and Ki-67 epitope as markers (59). In line with the slow growth of these lesions, cell proliferation was found to be modestly increased compared to the surrounding tissue (59). Malignant AFTNs are uncommon. In a study of 306 patients presenting with AFTNs, Horst et al. did not find any thyroid malignancy (5). Sandler et al. concluded that most reported hyperfunctioning carcinomas resulted from the coexistence of small malignancies in or adjacent to a benign hot lesion (14). Isolated cases of carcinomas in hot nodules have, however, been reported (4, 60-62). Smith et al. reported the occurrence of 3 carcinomas in 30 consecutive patients operated for solitary hot nodules (62). In a series of 164 patients, 3 of 29 patients treated surgically were diagnosed with thyroid cancer (63). It is an open question whether the diagnosis of cancer would be established in all these cases using modern histological criteria and molecular markers (64). In most instances, the presence of an AFTN argues against the presence of a malignant lesion.
TOXIC MULTINODULAR GOITER
Definition
Hyperthyroidism may occur due to AFTNs in a multinodular thyroid gland and this is discussed in detail in Chapter 17.
Clinical presentation
In addition to the signs and symptoms associated with hyperthyroidism, patients with large toxic multinodular goiters may also have dysphagia, shortness of breath, stridor, or hoarseness.
Diagnosis
The diagnostic approach is in general similar to patients with a solitary AFTN, but cross-sectional imaging with computer tomography and pulmonary function tests need to be considered in a subset of patients in whom compression by the goiter is evident or suspected.
Treatment
Therapeutically, surgery and radioiodine therapy are the most commonly used therapeutic modalities.
Pathogenesis
While the mechanisms underlying the development of nodules are of complex nature (65), it has become apparent that hyperfunctioning adenomas within multinodular goiters or autonomous areas within euthyroid goiters may also harbor somatic gain-of-function mutations in the TSH receptor (66-68). It is noteworthy that the mutations may differ among the adenomas within the same multinodular goiter (66). This observation is consistent with studies demonstrating distinct clonal origins of different thyroid adenomas within the same multinodular goiter (69). For example, in two adenomas from the same goiter, one neoplasm harbored a M453T mutation, the second adenoma a T632I substitution (66). In another study, L632I and F631L mutations were found in two distinct lesions within the same goiter, whereas another patient had two distinct toxic nodules with the same I630L mutation (67). These studies reveal that the pathogenesis of hyperfunctioning adenomas does not differ between solitary toxic adenomas and multinodular goiters. In a study analyzing hyperfunctioning and nonfunctioning areas from patients with toxic multinodular goiters, gain-of-function TSH receptor mutations were detected in 14 of 20 hyperfunctioning areas, whereas no mutation was identified in nonfunctioning nodules (70). On microscopic analysis, only two of the hyperfunctioning areas corresponded to classic adenomas surrounded by a capsule, whereas the remainder had the characteristic features of hyperplastic lesions. The development of multinodular goiters had been associated with a D727E germline polymorphism in the TSH receptor (71), but this finding could not be corroborated in other studies (72, 73). Constitutively activating TSH receptor mutations have also been detected in autoradiographically hyperfunctioning areas of goiters from euthyroid patients (74). The observation that TSH receptor mutations are rare in nonfunctioning adenomas, even if of monoclonal origin (68, 75), indicate that distinct mechanisms must be implicated in the abnormal growth leading to nonfunctioning nodules (65).
HYPERTHYROID THYROID CARCINOMA
Definition and Epidemiology
As discussed above, hyperfunctioning nodules are most commonly benign. Rarely, follicular carcinoma is associated with thyrotoxicosis. Ehrenheim compiled 20 such cases in 1986 (76), and Salvatori et al. reviewed 54 similar cases reported in the literature (77). Age and sex distribution in these patients does not differ from that of patients with follicular carcinoma without thyrotoxicosis.
Clinical presentation
Most commonly, thyrotoxicosis and thyroid carcinoma are diagnosed at the same time because of signs of thyrotoxicosis and the finding of a thyroid nodule prompting fine needle aspiration.
Diagnosis
Patients with thyrotoxic thyroid cancer have predominantly T3 thyrotoxicosis (78). Thyroglobulin levels are elevated. Cytology reveals most commonly follicular thyroid cancer (79), but hyperfunctinoning papillary thyroid cancer has also been documented (80). In patients who are on levothyroxine substitution after total thyroidectomy, the presence of hyperfunctioning metastases may not be readily apparent. Gradual reduction or withdrawal of levothyroxine therapy is necessary in order to recognize whether the thyrotoxicosis is caused by excessive exogenous levothyroxine or hyperfunctioning metastases. Whole-body scanning with radioiodine is used for the localization of the hyperfunctioning metastases.
Treatment
Treatment of patients with functioning thyroid carcinomas does not differ from the therapy of thyroid cancer patients without thyrotoxicosis, but appropriate control of the hyperthyroid state with antithyroid drugs and beta-blockers is important before submitting a patient to thyroid surgery or 131iodine therapy. Exacerbation of the hypermetabolic state with precipitation of thyroid storm has been reported in a patient undergoing radioiodine therapy for metastatic thyroid carcinoma without prior control with antithyroid drugs (81).
Pathogenesis
In well-differentiated thyroid cancers, mutations in the Gs alphA subunit and the TSH receptor genes occur only very rarely (36, 80, 82-86). Although constitutive activation of the cAMP pathway results in enhanced growth, it is not thought to be sufficient for malignant transformation of otherwise normal thyrocytes. A few patients with hyperthyroidism due to autonomously functioning thyroid cancers harboring mutations in the TSH receptor have, however, been identified. For example, Russo et al. reported a patient who presented with hyperthyroidism and increased uptake in two nodules, but suppressed uptake in the remainder of the gland (80). After surgical removal of the right thyroid lobe, histological examination revealed the presence of an insular papillary carcinoma with lymph node and lung metastases. Mutational analysis of the TSH receptor gene documented a somatic mutation, D633H, in DNA isolated from the primary tumor and metastatic tissue. Another mutation that activates both the cAMP and the IP3 pathways, I486F, was found in a hyperfunctioning well-differentiated follicular carcinoma in a patient presenting with hyperthyroidism and increased radioiodine uptake within the thyroid mass (86). It is conceivable that concomitant activation of these two signaling casca des may promote transformation. In a patient with an Hurthle cell carcinoma, Russo et al. identified a L677V TSH receptor mutation (85). Basal cAMP levels were increased in transfected Chinese hamster ovary (CHO) cells, but IP3 accumulation has not been determined. A somatic M453T substitution has been identified in a 11-year-old girl with a hyperfunctioning nodule and a papillary carcinoma (87). The same mutation has been found in the germline of two patients with congenital hyperthyroidism, but there was no suggestion that it is oncogenic (88, 89). Interestingly, however, overexpression of the M453T TSH receptor mutation in the FRTL-5 rat cell line was sufficient to induce neoplastic transformation as assessed by growth in semisolid medium and athymic mice (90). Follicular carcinomas have also been reported in patients with Graves' disease (91-93). It has been suggested that long-standing stimulation through TSH receptor-stimulating antibodies may play a role in the pathogenesis of these neoplasias (94). Whether thyroid carcinomas affecting patients with underlying Graves' disease behave more aggressively, as suggested by some authors (95), remains uncertain (96).
FAMILIAL NON-AUTOIMMUNE HYPERTHYROIDISM WITH TSHR MUTATIONS
Definition and Epidemiology
Autosomal dominant familial hyperthyroidism without evidence of an autoimmune etiology has been first described by Thomas et al. in 1982 (Chapter 16 A) (97). Currently 27 families with a total of 152 affected individuals with non-autoimmune familial hyperthyroidism have been reported (For recent review see: (98)). The hyperthyroidism is caused by monoallelic gain-of-function germline mutations in the TSH receptor.
Clinical presentation
The typical signs associated with autoimmune hyperthyroidism, i.e. the presence of stimulatory TSH receptor antibodies, endocrine ophthalmopathy, myxedema, lymphocytic infiltration of the thyroid gland, are absent. The age of onset of hyperthyroidism is variable and depends, in part, on the activity of the mutated allele. The majority of patients has a goiter.
Diagnosis
Affected individuals have a suppressed TSH and elevated peripheral hormones in the absence of TSH receptor-stimulating antibodies and TPO antibodies. The family history is key in order to demonstrate familial clustering suggestive for an autosomal dominant disorder. Ultimately, the diagnosis requires sequence analysis of the TSH receptor gene in order to evaluate it for the presence of a monalllelic mutation. If the mutation is unknown, functional in vitro analyses are needed to demonstrate that the mutated allele confers constitutive activity to the receptor.
Treatment
In order to achieve permanent cure, it is necessary to destroy all thyroid tissue, either by thyroidectomy followed by radioiodine therapy, or radiotherapy alone (98). In younger patients, temporary therapy with thionamides can be considered. Because the condition may not be readily recognized and confused with Graves' disease, patients treated with thionamides or insufficient amounts of radioiodine have frequent relapses (98).
Pathogenesis
The molecular basis of hereditary non-autoimmune hyperthyroidism was elucidated by detecting activating germline mutations in the TSH receptor in the family reported by Thomas et al. (51). Gain-of-function mutations are by definition dominant, and alteration of one allele is thus sufficient for generating the phenotype. Interestingly, the onset of hyperthyroidism may vary in carriers of the same mutation in a given kindred. Hence, other factors, for example genetic background and/or iodine intake, appear to modulate the phenotypic expression (97, 99, 100).
SPORADIC NON-AUTOIMMUNE HYPERTHYROIDISM
Autoimmune neonatal hyperthyroidism is rare and occurs in less than 2% of newborns that are the offspring of a mother with a history of Graves' disease (101), a condition with an estimated incidence of about 2 of every 1000 pregnancies (97). In these infants, the congenital hyperthyroidism is caused by transplacental passage of stimulating TSH receptor autoantibodies (102). Antibody-induced neonatal hyperthyroidism usually resolves within the first few months of life as the maternal antibodies are cleared from the circulation. Occasionally, thyroid hormone may be fluctuating between elevated and decreased levels because of the concomitant presence of stimulating and blocking antibodies (103). Constitutively activating germline neomutations in the TSH receptor have been found in a total of 15 patients with sporadic congenital non-autoimmune hyperthyroidism (Chapter 16 A) (104)(For recent review see:(98)). Congenital hyperthyroidism due to a toxic adenoma harboring a somatic TSH receptor mutation was reported as an unusual variant (8). The patients with non-autoimmune congenital hyperthyroidism must be differentiated from the much more common and transient autoimmune form of neonatal hyperthyroidism, because these patients have pronounced hyperthyroidism requiring a more aggressive therapeutic approach that may necessitate surgery and ablative radiotherapy early in life. Several of the children with severe neonatal hyperthyroidism were reported to have mild mental retardation (104-106), suggesting that high levels of thyroid hormone may have a negative impact on brain development (107). Alternatively, mental development may have been impaired because of premature closure of the cranial sutures. A subset of these children had proptosis (88, 89). Computer tomography of the retroorbital tissue in one of these children did, however, not demonstrate infiltration of the eye muscles (88).
TSH-SECRETING PITUITARY ADENOMA
Definition and Epidemiology
TSH-secreting adenomas (TSHomas) account for less than 2% of all pituitary adenomas and are a rare cause of thyrotoxicosis (Chapter 13) (108, 109). TSHomas and RTH form the two syndromes of "inappropriate TSH secretion", defined by normal or elevated TSH levels in combination with increased (free) T4 and T3 levels.
Clinical presentation
Patients with TSHomas present with signs and symptoms of hyperthyroidism and an enlarged thyroid. In patients with RTH, the phenotype is more complex as some tissues are resistant to the action of the elevated peripheral hormones and thus hypothyroid, whereas other tissues can be excessively stimulated. The physiological negative feedback normally exerted by thyroid hormones is not operating in both conditions. TSHomas secrete TSH in an autonomous fashion, in RTH the thyrotropes are resistant to the high levels of thyroid hormone. Most patients are older, but TSHomas have also been documented in children (110).
Diagnosis
Magnetic resonance imaging (MRI) of the pituitary will reveal a pituitary adenoma in patients with TSHomas. TSH is formed of a specific beta subunit and the glycoprotein alpha subunit common to TSH, FSH and LH/CG. Some clinicians measure the glycoprotein alpha subunit as a marker to distinguish between TSHomas and RTH. The alpha subunit and the beta subunit /TSH ratio are often elevated in patients with TSHomas (111). However, in one series of TSHomas, normal alpha subunit levels were observed in more than 60% of the patients, particularly in microadenomas (112). The TSH secreted by TSHomas is normal in terms of amino acid sequence, but has variable biological activity and is secreted in fluctuating amounts (113). Compared to controls, TSH burst frequency and basal secretion are increased, TSH secretion patterns are more irregular, but the diurnal rhythm is preserved at a higher mean in all patients (114). A more sensitive and specific test than measuring the alpha subunit consists in the T3 suppression test (80-100 µg of T3 per day per 8-10 days), which does not result in complete inhibition of T3 secretion in patients with TSHomas (108, 112, 115). An alternative consists in the TRH-stimulation test, but TRH is currently not available in the United States. After injection of TRH (200 µg i.v.) TSH and the alpha subunit do not increase in patients with TSHomas (108).
Treatment
Transsphenoidal surgery is the cornerstone for therapy of TSHomas. Complete resection may not be possible because these tumors can invade the sinus cavernosus and other adjacent structures. Prior to surgery, the hyperthyroidism should be controlled with thionamides and beta-blockers. In patients with residual tumor tissue and persistent secretion of TSH, both -knife radiotherapy (10 � 25 Gy) and medical therapies can be considered. The latter include the use of somatostatin analogues, such as octreotide and lanreotide. TSHomas express somatostatin receptors and somatostatin analogues are highly effective in reducing TSH secretion by neoplastic thyrotropes (116). If tolerated, somatostatin analogues are effective in reducing TSH secretion in more than 90% of patients with consequent normalization of thyroid hormone levels and restoration of the euthyroid state. Tumor shrinkage does occur in about 45% of patients (117). Dopamine receptors are also present in TSHomas and dopamine agonists such as bromocriptine or cabergoline have been used in order to control TSH secretion (108). The response is, however, highly heterogeneous and best in tumors secreting both TSH and prolactin. In the case of a surgical cure, the postoperative TSH is undetectable and may remain low for weeks or months, causing central hypothyroidism. Permanent central hypothyroidism may also occur due to the mass effect exerted by the tumor or after radiotherapy. Thus, transient or permanent substitution therapy with levothyroxine may be necessary. Long-term evaluation of all pituitary axes is important, particularly in patients who underwent radiotherapy, in order to recognize and treat anterior pituitary deficiencies in a timely manner.
Pathogenesis
The molecular mechanisms leading to the formation of TSHomas remain unknown. TSHomas have been shown to be monoclonal by X-inactivation analyses suggesting that they arise from a single cell harboring one or several mutations in genes controlling proliferation and perhaps function (108).
RESISTANCE TO THYROID HORMONE
The syndrome of Resistance to Thyroid Hormone (RTH) is described in detail in Chapter 16 D. RTH is defined by elevated circulating levels of free thyroid hormones due to reduced target tissue responsiveness and normal, or elevated, levels of TSH (118, 119). Patients with RTH typically present with goiter. Their metabolic state may appear euthyroid or include signs of hypo- and hyperthyroidism. With the exception of the first studied kindred, a single sibship harboring a deletion of the entire coding sequence of the entire TRbeta gene and a recessive pattern of inheritance, RTH is most commonly caused by monoallelic mutations of the TRbeta gene. The mutation can be inherited in an autosomal dominant manner or occur as de novo mutation. The mutant receptors act in a dominant negative fashion to block the activity of the normal allele, thereby explaining the dominant inheritance. The gene defect remains unknown in about 15% of subjects with a RTH phenotype. It is likely that mutations in cofactors that are required for normal TR function are involved in the pathogenesis of RTH in these patients. The generation of mice with targeted deletion of TRbeta, as well as TR knockin models, have been essential for elucidating the physiology of thyroid hormone action and the pathophysiology of RTH (120).
HCG-INDUCED GESTATIONAL HYPERTHYROIDISM
Thyrotoxicosis and other forms of thyroid dysfunction in the pregnant patient are discussed in detail in Chapter 14.
Definition and Epidemiology
Gestational transient thyrotoxicosis of non-autoimmune origin is caused by stimulation of the TSH receptor through hCG (121, 122). hCG-induced hyperthyroidism occurs in about 1.4 % of pregnant women, mostly when hCG levels are above 70-80,000 IU/l (123, 124) .
Clinical presentation
Many signs and symptoms of hyperthyroidism are not specific and overlap with those of normal pregnancy (125). Hence, the accuracy of clinical diagnosis is limited. Because of the decrease in the levels and bioactivity of hCG later in pregnancy, hCG-induced gestational hyperthyroidism is usually transient and limited to the first 3-4 months of gestation. In a subset of women, the manifestations of hCG-induced hyperthyroidism are more severe and they are often associated with hyperemesis. Goodwin et al. studied the relationship of serum hCG, thyroid function, and severity of vomiting among 57 hyperemesis patients and 57 controls matched for gestational age (126). Hyperemesis patients had significantly greater mean serum levels of hCG, free T4, total T3, and estradiol, and lesser serum TSH concentrations compared to controls. The degree of biochemical hyperthyroidism and hCG concentration correlated directly with the severity of vomiting. The hyperemesis may be caused by a marked hCG-induced increase in estradiol levels (122). However, the relation between hyperemesis and gestational hyperthyroidism varies among patients, and additional, unidentified mechanisms may be involved.
Diagnosis
The diagnosis is established by measuring TSH, free or total T4, and T3. The physiological decrease in TSH levels and the increase in total thyroid hormone concentrations associated with the increase in thyroxine-binding globulin (TBG) have to be considered when interpreting the results. TBG levels increase in response to elevated estradiol levels and plateau by about 20 weeks of gestation (127). Therefore, total T4 and total T3 levels increase by approximately 1.5 fold. If free T4 levels are determined by analogue assays, serum concentrations are usually significantly lower than values in non-pregnant women (128).
Treatment
Treatment with antithyroid medications is often not necessary. Women with hyperemesis need therapy with antiemetics. In patients in whom total T4 levels are higher than 1.5 times the upper reference range, therapy with antithyroid drugs may be indicated. Propylthiouracil (PTU) is the preferred medication during the first trimester and methimazole during the remainder of pregnancy in the United States (129, 130). A review by Mandel and Cooper has specifically addressed the use of thionamides during pregnancy and lactation (131). Overtreatment with antithyroid drugs can result in hypothyroidism in the fetus. Therefore, free T4 should be kept close or slightly above the normal range with the lowest possible dose of antithyroid drugs.
Pathogenesis
The pathophysiology of hCG-induced gestational thyrotoxicosis has been reviewed by Hershman (132). hCG and TSH share the common glycoprotein alpha subunit and the beta subunit is highly homologous. At high doses, hCG cross-reacts with the TSH receptor, and this stimulation can lead to an increase in secretion of T4 and T3, with subsequent suppression of TSH secretion (124, 133). The levels of hCG and TSH are inversely correlated during the first trimester (121). Free T4 levels determined between weeks 6-20 of gestation increase and show a linear relationship with the rising hCG levels (134). The thyroid gland of normal pregnant women may be stimulated by hCG to secrete slightly excessive quantities of T4 and induce a slight suppression of TSH, but it only induces overt hyperthyroidism in a subset of pregnant women. The increased secretion of hCG result only in the physiological decrease in TSH levels that are characteristic for the first trimester of pregnancy, or in overt hyperthyroidism. Of note, elevations of hCG are particularly pronounced in twin pregnancies (135). In a study characterizing the activity of hCG on the human thyroid gland, 1.0 U hCG was found to be roughly equivalent to 0.27 mU of TSH (136). LH also has intrinsic thyroid stimulating activity, but it is lower compared to hCG. TSH-binding and TSH-induced adenylyl cyclase stimulation are more effectively inhibited by desialylated variants of hCG than unmodified hCG (137). Nicked hCG preparations, obtained from patients with trophoblastic disease or by enzymatic digestion of intact hCG, showed approximately 1.5- to 2-fold stimulation of recombinant hTSH receptor compared with intact hCG (122). Deglycosylation and/or desialylation of hCG enhance its thyrotropic potency. Basic hCG isoforms with lower sialic acid content extracted from hydatiform moles were more potent in activating the TSH receptor. From these and other studies it seems that the biological effect of hCG is predominantly confined to hCG containing little or no sialic acid. hCG has also been found to increase iodide uptake in cultured FRTL-5 cells and it also causes a dose related increase of adenylyl cyclase activity and thymidine uptake (138, 139).
FAMILIAL HYPERSENSITIVITY TO HCG
An unusual form of familial gestational hyperthyroidism caused by a mutant TSH receptor displaying hypersensitivity to normal levels of hCG has been identified by Rodien et al. (140). The index patient had a history of two miscarriages that were accompanied by hyperemesis. Subsequently, she had two pregnancies that were complicated by hyperthyroidism, severe nausea and vomiting. She did not have any antibodies against the TSH receptor or TPO. Her hCG levels, determined during the second pregnancy, were in the normal range for the first trimester. The patient' s mother had a history of one miscarriage and two pregnancies that were complicated by hyperemesis gravidarum. Sequence analysis of the TSH receptor gene in the proband and her mother revealed the presence of a monoallelic point mutation resulting in the substitution of K183R. Functional studies in COS-7 cells transfected with the mutated receptor documented no differences in membrane expression, and similar levels of basal and TSH stimulated cAMP accumulation. In contrast to the wild-type TSH receptor, which reacts only minimally to high doses of hCG, the K183R mutant is hypersensitive to hCG, although it still is 1000 times less responsive to hCG than the LH/CG receptor. The K183R TSH receptor mutation is unique because sensitivity is increased for hCG but remains unaltered for the cognate ligand TSH (140). This observation also supports the possibility of an hCG-independent connection between hyperthyroidism and hyperemesis gravidarum.
TROPHOBLAST TUMORS: HYDATIFORM MOLES AND CHORIOCARCINOMA
Definition and Epidemiology
Gestational trophoblastic diseases comprise hydatiform moles, invasive moles, choriocarcinomas and placental site trophoblastic tumors (141). Hydatiform moles and choriocarcinomas that secrete high amounts of hCG can cause hyperthyroidism (142). In 1955 Tisne et al. described a patient with molar pregnancy that had increased thyroidal uptake of radioactive iodine and clinical signs of hyperthyroidism (143). Earlier reports also described molar pregnancies in combination with hyperthyroidism and in all cases a rapid return to normal thyroid function occurred after removal of the mole (143). In men, choriocarcinomas can arise in the testis and cause hyperthyroidism by secreting hCG (144). In a study of 20 patients with gestational trophoblastic neoplasias, 2 patients were overtly thyrotoxic and this was confirmed by elevated serum T4 levels (145). These 2 patients had extremely high serum (3,220,000 IU/l and 6,720,000 IU/l) and urine hCG levels, which correlated closely with TSH-like activity exerted by the serum of these patients in a mouse thyroid bioassay. Patients with moderately increased serum hCG levels (110,000-310,000 IU/l) associated with trophoblastic neoplasia were euthyroid. A similar correlation between serum hCG levels and thyroid stimulating activity in both serum and urine was found in women who had widely metastatic choriocarcinoma and marked hyperthyroidism (145). In another patient with gestational choriocarcinoma serum thyroid stimulating activity correlated precisely with serum T4, with the beta subunit of hCG, and with the quantification of the tumor burden (146). Hyperthyroidism associated with choriocarcinoma in the male is extremely rare, but has been reported repeatedly (122). Orgiazzi et al. compiled four cases from the literature and reported a patient who had choriocarcinoma of the colon associated with gynecomastia and hyperthyroidism (147). Thyroid stimulating activity, measured by a mouse bioassay, was detected in the serum. Serum thyroid stimulating activity was partly inactivated by antibovine-TSH antiserum, but was completely neutralized by anti-hCG antiserum.
Clinical presentation
Most women with hydatiform moles present with uterine bleeding in the first half of pregnancy. The size of the uterus is large for the duration of gestation (141). Many women with molar pregnancies have nausea and vomiting, some have pregnancy-induced hypertension or (pre)-eclampsia. The signs and symptoms of thyrotoxicosis are present in some women, but they may be obscured by toxemic signs. The characteristic features belonging to Graves' disease are missing. The thyrotoxicosis is usually not severe because of a relatively short duration. Women with choriocarcinomas present within one year after conception. The tumor may be confined to the uterus, more frequently it is metastatic to multiple organs such as the liver and lungs, among others. In men, choriocarcinomas of the testes is often widely metastatic at initial presentation. Gynecomastia is a common finding.
Diagnosis
Measurement of serum hCG concentrations is needed for the diagnosis of moles and choriocarcinomas, and hCG serves as a sensitive and specific tumor marker during therapy and surveillance (145). In women, hCG concentrations are significantly higher than those found during normal pregnancies. Ultrasonography of the uterus shows a characteristic "snowstorm" pattern. The diagnosis of thyrotoxicosis relies on the measurement of TSH, (free) T4 and T3. elevated thyroid hormone levels. Thyroidal radioiodine uptake is elevated.
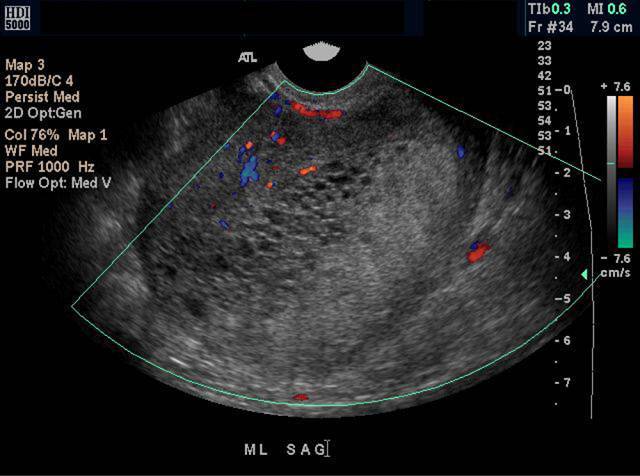
Treatment
Hydatiform moles are treated by suction rather than curettage (148). Serum T4, T3, TSH, and hCG levels normalize rapidly after removal of the mole. Choriocarcinomas can be divided into two groups: 1) a low risk group treated by monotherapy, most often with methotrexate or actinomycine D and a success rate close to 100%, and 2) a high risk group treated with polychemotherapy (etoposide, methotrexate, actinomycine D, cyclophosphamide, vincristine) with a response of about 86%. In patients that are not responding to chemotherapy, the 5-year survival rate is about 43%. Longitudinal measurement of hCG as specific and sensitive tumor marker is key for long-term surveillance (148). Placental-site trophoblastic tumors, a rare form of gestational trophoblastic disease that does not secrete hCG, requires stage-adapted management with surgery, or surgery in combination with chemotherapy (149).
STRUMA OVARII
Definition and Epidemiology
Struma ovarii is a rare tumor consisting primarily of thyroid components occurring in a teratoma or dermoid in the ovary (150). It forms less than 1% of all ovarian tumors and 2 to 4 % of all ovarian teratomas; 5 to 10% are bilateral, and 5 to 10% are malignant (151, 152). Thyrotoxicosis occurs in about 8% of affected patients (153).
Clinical presentation
The clinical presentation may include the finding of an abdominal mass, ascites, pelvic pain, and, rarely, a pseudo-Meigs syndrome with pleural effusions (154). A subset of women present with subclinical or overt thyrotoxicosis. Goiter is only presented in patients with associated thyroid disease. For example, coexistence of Graves' disease and struma ovarii has been reported (155).
Diagnosis
In patients with thyrotoxicosis, TSH is suppressed and T3 and T4 levels are elevated. Thyroglobulin is secreted by benign and malignant ovarian strumae. Radioiodine uptake will reveal uptake in the pelvis, while the uptake in the thyroid is diminished or absent (156). Cross-sectional imaging with computed tomography or magnetic resonance imaging will demonstrate of uni- or bilateral ovarian masses (156). CA125 may be elevated (154). Malignant thyroid tissue shows the characteristic patterns of papillary or follicular thyroid cancer and can be positive for mutations in BRAF (157). Metastasis is uncommon but has been reported repeatedly (158).
Treatment
Unilateral or bilateral open or laparoscopic oophorectomy is the primary therapy (159). Thyrotoxic women should be treated with antithyroid drugs and, if needed, with beta-blockers prior to surgery. In the case of malignant lesions, the patient should undergo thyroidectomy followed by treatment with 131iodine (157). The subsequent surveillance for residual or recurrent thyroid cancer does not differ from primary thyroid carcinomas.
IODINE-INDUCED HYPERTHYROIDISM
I. IODINE-INDUCED THYROTOXICOSIS
Definition and Epidemiology
An excess of iodine through dietary intake, drugs or other iodine-containing compounds can lead to thyrotoxicosis through increased thyroid hormone synthesis in the presence of underlying thyroid disease, particularly multinodular goiters that contain zones of autonomy (160, 161). Iodine-induced thyrotoxicosis (IIT) has been recognized as early as 1821 by Coindet, who reported that goitrous individuals treated with iodine developed hyperthyroidism (162). The condition is now commonly called Jod-Basedow (Jod = iodine in German; Karl von Basedow = German physician describing the signs of thyrotoxicosis associated with exophthalmos and goiter, i.e. Graves' disease) (163). IIT may occur in patients from endemic goiter areas, patients with multinodular goiters in non-endemic areas, individuals with Graves' disease, and in individuals without previously apparent thyroid disease (164). The sources of iodide leading to IIT are manifold (Table 13-2). IIT has been reported after initiating iodine supplementation, but also with the use of iodinated drugs, contrast agents, and food components (165, 166). Use of non-ionic contrast agents does not prevent the development of IIT (167).
Table 13-2 Iodine-containing compounds potentially associated with IIT
| Radiological contrast agents |
| Diatrizoate |
| Ipanoic acid |
| Ipodate |
| Iothalamate |
| Metrizamide |
| Diatrozide |
| Topical iodine preparations |
| Diiodohydroxyquinolone |
| Iodine tincture |
| Povidone iodine |
| Iodochlorohydroxyquinolone |
| Iodoform gauze |
| Solutions |
| Saturated potassium iodide (SSKI) |
| Lugol solution |
| Iodinated glycerol |
| Echothiopate iodide |
| Hydriodic acid syrup |
| Calcium iodide |
| Drugs |
| Amiodarone |
| Expectorants |
| Vitamins containing iodine |
| Iodochlorohydroxyquinolone |
| Diiodohydroxyquinolone |
| Potassium iodide |
| Benziodarone |
| Isopropamide iodide |
| Food components |
| Kelp, Kombu and other algae |
| Food colors: Erythrosine |
| Iodine containing food: Hamburger thyroiditis |
For adults, the Dietary Reference Intake for iodine is 150 μg (168). The Tolerable Upper Intake Level for adults has been set to 1,100 μg/day and was assessed by analyzing the effect of supplementation on TSH (169). The thyroid gland needs no more than 70 μg/day to synthesize the required daily amounts of T4 and T3 (170). The higher recommended daily allowance (RDA) levels of iodine are recommended for optimal function of a number of organs such as the lactating breast, gastric mucosa, salivary glands, oral mucosa, thymus, epidermis, and the choroid plexus. The normal thyroid protects itself from acute excessive amounts of iodide by the Wolf-Chaikoff effect, which consists of an immediate reduction in iodide uptake, iodide organification, thyroid hormone biosynthesis and secretion (171). Remarkably, most individuals with a normal thyroid gland also tolerate a chronic excess of 30 mg up to 2 g iodide per day without clinical symptoms (161). Thyroid function tests remain within the reference range although T4 and T3 drop, and TSH rises (161). However, in some individuals, even exposure to modest amounts of excessive iodine can induce IIT or hypothyroidism. Fears that iodine supplementation would lead to IIT led to opposition against iodination programs in Switzerland, but initiation of salt supplementation with very low doses of iodine (3.75 parts per million) were shown to be safe (172). This contrasts with the observations from other iodination programs using higher amounts of iodide. For example, a steep rise of IIT has also been documented in 1966 in Tasmania (Australia), an area of iodine deficiency with a high prevalence of goiter (173). This was associated with the addition of potassium iodide to bread in early 1966. The increased incidence occurred predominantly in subjects older than 40 years, in whom a rise in incidence from 50 to a maximum of 130 cases per 100,000 was seen between 1967 and 1968. By 1974, the incidence decreased to the pre-epidemic level. Most thyrotoxic patients had nodular goiters and few patients had underlying Graves' disease. Later it was recognized that there was already a pre-epidemic increase in the incidence of thyrotoxicosis caused by the use of iodofor disinfectants on dairy farms (174, 175). Recent supplementation programs using inadequately high amounts of iodide in endemic goiter regions in Zimbabwe and eastern Zaire also resulted in a significant number of cases of severe and long-lasting IIT (176, 177). Three years after starting supplementation with iodized salt in China, the prevalence of overt hyperthyroidism in three cohorts was 1.6%, 2.0% and 1.2%, irrespective of the nutritional iodide intake, which varied between mildly deficient, adequate or excessive ((178, 179). The incidence of IIT during the first three years of supplementation was not determined. In these three communities, the cumulative 5-year incidences of overt hyperthyroidism for the 4th to 8th year of supplementation were 0.4%, 1.2% and 1.0%. At first glance, this seems to indicate a very low risk of IIT. However, the calculated 1-year incidence rates are 80, 240 and 200 per 100,000 individuals per year, figures that are much higher than the incidence rates published in other countries (180).
In some patients, iodine excess causes overt clinical hypothyroidism. Patients with a history of Graves' disease treated with radioiodine or partial thyroidectomy, partial thyroidectomy for thyroid nodules, or autoimmune thyroiditis appear to be particularly predisposed to iodine-induced hypothyroidism (181-184). Even relatively small excessive doses of 750 ug may be sufficient to induce hypothyroidism (185).
Clinical presentation
The clinical presentation includes the typical signs of thyrotoxicosis and in most patients the finding of a multinodular goiter. Other patients may have underlying autoimmune thyroid disease. A pre-existing thyroid disorder is been present in at least 20% of patients.
Diagnosis
The diagnostic considerations are the same as for toxic nodular goiters.
Treatment
Spontaneous reversal to an euthyroid state may occur after a mean period of 6 months in about 50% of patients. Return to euthyroidism may be preceded by subclinical hypothyroidism (186). In patients with multinodular goiters, the therapeutic considerations are the same as discussed for toxic nodular goiters.
Pathogenesis
In a classical study, four euthyroid patients with a single autonomous nodule from the slightly iodine-deficient Brussels region received a supplement of 500 μg iodide per day (187). This caused a slow but constant increase of thyroid hormone. After four weeks, the patients became hyperthyroid . Later studies confirmed the original interpretation that the nodules were not producing excessive amounts of thyroid hormones because of the low iodine intake, but that they became toxic once presented with high amounts of iodine (188). The autonomy of function was secondary to gain-of-function mutations in the TSH receptor (54). Individuals with multinodular goiters living in iodine-replete regions can also develop hyperthyroidism, albeit at much higher doses of iodine (up to 180 mg) (189). Taken together, these observations confirm that individuals with (multi)nodular goiters are particularly prone to developing IIT (161). In regions with iodine deficiency, nodular goiters disappear slowly after the introduction of iodine supplementation and the incidence of hyperthyroidism then gradually decreases over the years (190, 191). More recent data suggest that autonomous nodules are not the only explanation for the pathogenesis of IIT. Several human and animal studies suggest that chronic excessive iodine intake may modulate thyroid autoimmunity and lead to thyrotoxicosis in genetically susceptible individuals. Mice prone to developing autoimmune disease that are first fed an iodine-deficient diet and then switched to iodine excess develop dose-dependent ultra-structural changes in thyrocytes that are consistent with autoimmune disease (192). A necrotic effect of excessive amounts of iodide has been demonstrated in vivo in various animal species and also in human thyroid follicles in vitro (193). Epidemiologic studies performed in China, Turkey and Denmark suggest that supplementation with iodized salt increases the prevalence of autoimmune thyroid disease, resulting in clinical or subclinical hypothyroidism (178), autoimmune hyperthyroidism (194), or both (195). In a population-based, cross-sectional study with 1085 participants exposed to excessive amounts of iodide (40-100 mg/kg salt) from Brazil, the prevalence of chronic autoimmune thyroiditis was 16.9%, women were more commonly affected (21.5 versus 9.1%), 8% were hypothyroid, and 3.3% were hyperthyroid (196). The authors concluded that the excessive iodine may have increased the prevalence of autoimmune thyroid disease and hypothyroidism in this population (196). In Denmark, a moderately iodine-deficient country, introduction of iodized salt at a dose that was calculated to increase iodine intake by only 50 ug per day, an increased incidence of hyperthyroidism was found mainly in younger patients between the age of 20 and 39 years and was presumably induced by autoimmune thyroid disease (194). In contrast, a study on children in Morocco did not find an effect of iodine supplementation on thyroid autoimmunity (197). It is noteworthy that IIT was also observed in individuals who probably had normal thyroid glands (164, 186, 198). It is well recognized that contrast agents may cause IIT. They contain between 30-50% of iodine and several grams are used freqeuently for radiological studies. Individuals with multinodular goiters or subjects who live in countries where iodine intake is low are at particular risk for developing IIT secondary to exposure to contrast agents (160). Clinicians should be aware that IIT often develops several weeks after their administration. Follow-up of such patients after radiological procedures is therefore advisable and in some cases prophylactic therapy with methimazole prior to the administration of contrast agents may be indicated. Considering the wide use of contrast agents, the probability of inducing IIT by these substances appears to be relatively low. However, the incidence of IIT appears to be inversely related to the nutritional iodine intake.
II. AMIODARONE INDUCED THYROID DISEASE
Definition and Epidemiology
Amiodarone is a widely used anti-arrhythmic agent used for the therapy of ventricular and supraventricular arrhythmias, among them atrial fibrillation. It has a very high iodine content of 37.3% by weight. About 3 mg of iodine are released into the circulation per 100 mg of amiodarone ingested. For comparison, the RDA for iodide is 150 μg per day. Its molecular structure has some similarities with iodothyronines and it may interfere with thyroid hormone transport into cells and with intracellular thyroid hormone metabolism and action (Figure 13-2) (199, 200). Amiodarone interferes with 5'-monodeiodination of thyroid hormones leading to a decrease of both intra- and extracellular T3 concentrations.
Amiodarone-induced hyper- and hypothyroidism play an important role in clinical practice (200, 201). Amiodarone-induced thyrotoxicosis (AIT) is more common in iodine-deficient regions, but also occurs in patients with a normal nutritional iodine intake. Amiodarone-induced hypothyroidism is usually seen in iodine-sufficient areas.
Reported incidences of AIT vary between 0.003% and 11.5%. In a study involving 1448 patients treated with amiodarone, 30 developed AIT (164). AIT is differentiated into two forms. AIT Type I is caused by increased hormone synthesis because of exposure to high amounts of iodine, AIT Type II results from cytotoxic destruction of thyrocytes (200). Hypothyroidism occurs predominantly in patients with preexisting thyroid autoimmune disease and in areas of normal iodine intake (199, 202). AIT is more common in men than in women (203).
Clinical presentation
The signs of thyrotoxicosis are not apparent in all patients with AIT. They may be obscured by the underlying cardiac condition. Some patients have a nodular goiter.
Diagnosis
The total or free T4 levels are elevated in euthyroid, hypothyroid and hyperthyroid patients treated with amiodarone because of its inhibition of 5'-monodeiodination. In hyperthyroid patients, TSH is suppressed and T3 is elevated. The distinction between AIT Type I and II can be difficult on clinical grounds. The radioiodine uptake is typically low to normal in AIT Type I, and low to suppressed in AIT Type II. Serum interleukin 6 levels are normal to high in AIT Type I and markedly elevated in AIT Type II, but there is significant overlap and the test is of insufficient sensitivity. On Doppler ultrasound, AIT Type I is associated with normal to increased vascularity with patchy distribution, while Type II shows absent vascularity (204, 205).
Treatment
The therapy of AIT is a challenge. An algorithm for the management of patients with AIT is shown in Figure 13-3 (200). If possible, amiodarone should be discontinued. Patients with type 1 AIT are preferably treated with methimazole (initially 40-60 mg/day, followed by gradual adjustment of the dose), but the response to thionamides is modest. In selected patients, treatment with potassium perchlorate (1 g/day for 4 to 6 weeks) can be considered. Potassium perchlorate is a drug that can cause aplastic anemia and its use should be limited to patients who cannot be controlled by methimazole, or who are allergic to thionamides. For patients with AIT Type II, prednisone (0.5 - 0.7 mg /kg body weight per day) can be used for several months. Because the distinction between AIT Type I and II is difficult and not always clear, and because some patients have mixed forms of AIT, these therapies are occasionally combined. Patients with a history of AIT type II are at risk for developing hypothyroidism if exposed to high amounts of iodide.
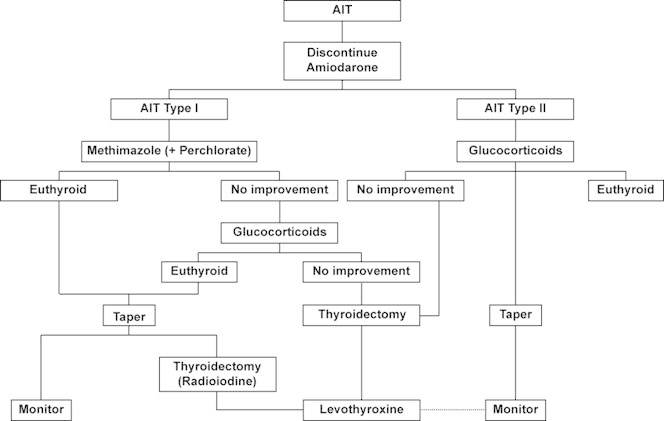
Pathogenesis
Two distinct mechanisms result in AIT. AIT Type I results from the iodine-induced increase in thyroid hormone synthesis. Patients developing AIT Type I usually have a preexisting nodular goiter. AIT Type II is caused by cytotoxic effects of the medication that results in the release of preformed thyroid hormones.
Pathology
On electron microscopy imaging, AIT Type II shows characteristic multilamellar lysosomal inclusions and intramitochondrial glycogen inclusions, and a morphological picture of thyrocyte hyperfunction (206). No inflammatory changes are present.
THYROIDITIS
Any form of thyroiditis can be associated with a thyrotoxic phase because the disruption of thyroid follicles can result in an increased release of stored iodothyronines. The thyrotoxic phase may be followed by transient or permanent hypothyroidism.
I. ACUTE OR SUBACUTE (DE QUERVAIN'S) THYROIDITIS
Definition
This disorder, which is discussed in Chapter 19, leads to temporary thyrotoxicosis in approximately half of the patients due to discharge of stored hormone from the thyroid gland.
Clinical presentation
Patients with subacute thyroiditis often present with a history of a preceding respiratory tract infection (207). They may have fever, malaise, and soreness, and the gland is exquisitely tender on palpation and often displays a substantially increased consistency.
Diagnosis
The laboratory findings will fluctuate with the course of the disease and typically present with initial thyrotoxicosis, followed by a hypothyroid phase. The thyroid function may normalize or result in permanent underfunction. The erythrocyte sedimentation rate is markedly elevated. Thyroid antibodies are usually not detectable. The radioiodine uptake is extremely low or absent. Thyroglobulin levels are elevated because of the destruction of thyroid follicles.
Treatment
Symptomatic treatment with non-steroidal anti-inflammatory drugs (NSAID) or aspirin is often sufficient. A subset of patients needs therapy with prednisone for variable amounts of time. Addition of a beta-blocker should be considered based on the severity of the thyrotoxic signs. Therapy with levothyroxine may be necessary during the hypothyroid phase of the illness. While the majority of patients recover completely, about 10% of cases develop permanent hypothyroidism.
Pathogenesis
The pathogenesis is thought to involve a viral infection.
Pathology
Cytology and histology show characteristic giant cells.
II. SILENT OR PAINLESS THYROIDITIS
Definition and Epidemiology
Silent thyroiditis is characterized by lymphocytic infiltration and can lead to thyrotoxicosis and hypothyroidism (208). Although the terms silent thyroiditis and painless thyroiditis are used most commonly, many other names have been used for this disorder including sporadic thyroiditis (208), destructive thyroiditis (209), hyperthyroiditis (210), spontaneously resolving lymphocytic thyroiditis (211), transient painless thyroiditis (212), painless thyroiditis with transient hyperthyroidism (213), painless subacute thyroiditis (214), occult subacute thyroiditis (215), atypical thyroiditis (216) and transient thyrotoxicosis with lymphocytic thyroiditis (217). Silent thyroiditis has been diagnosed frequently in the 1970s, but its incidence seems to be lower now. A retrospective survey conducted in Wisconsin from 1963 through 1977 showed that silent thyroiditis was not found until 1969 and was uncommon up to 1973 (212). The frequency then increased and silent thyroiditis was thought to be responsible for about 20% of all cases of thyrotoxicosis in this geographical area (211). This high incidence has not been reported in other regions of the United States, Asia and Europe. In a study from Japan, an incidence of 10% was found in the 1980s, but in New York it was only 2.4% (218). Schneeberg reported data obtained from a random poll; the obtained data suggest that silent thyroiditis was uncommon in Argentina, Europe and the East- and the West coast of the United States, but occurred more frequently around the Great Lakes and in Canada (219). The variable incidence rates may be due to an ascertainment bias or to the development of thyrotoxicosis secondary to the ingestion of meat contaminated with bovine thyroid tissue. Two epidemics of thyrotoxicosis thought to reflect silent thyroiditis were found to be explained by meat contamination (220, 221). Affected patients are mostly between 30 and 60 years of age and the female to male ratio is about 1.5: 1 (211). The condition is currently rarely recognized.
Clinical presentation
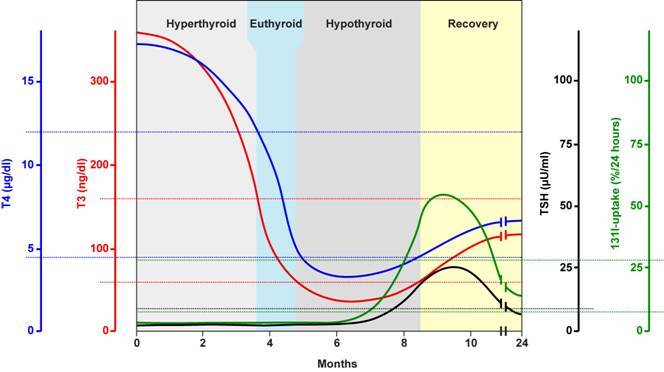
Patients present with abrupt onset of thyrotoxicosis that can be associated with the development of a goiter or enlargement of a preexisting goiter. Repeated episodes may occur in the same individual (211). In a review on 112 patients, 68 were female and the age at onset was 32.4 +/- 18.5 in females and 24.9 +/- 8.2 years in males (213). None of the patients presented with thyroid pain. The duration of the thyrotoxic phase was variable, but for the most part, it lasted less than one year. The mean duration was 3.6 months (range 1-12.5). Symptoms began 2.5 - 2.2 months preceding the initial evaluation. This period is shorter than is usually seen with Graves' disease and much shorter than in patients with toxic multinodular goiter. Exophthalmos and pretibial myxedema were absent. The thyroid gland is typically firm in consistency. Forty three percent of patients had an enlarged thyroid, which was generally symmetrical and enlargement was in most instances mild. The clinical course of the disease consists usually of an initial hyperthyroid phase, followed by a hypothyroid phase, and subsequent restoration of a euthyroid metabolic state (Figure 13-4). 57 out of 112 patients became euthyroid and did not develop clinical hypothyroidism. After a brief period of euthyroidism, transient biochemical hypothyroidism developed in 17 patients. In 32 patients clinical hypothyroidism was present (213).
Development of Graves' disease, after painless thyroiditis has been documented and TSH receptor antibodies have been found in these patients (222).
Diagnosis
During the first phase of the disease, discharge of hormone from the inflamed thyroid results in increases in serum T4, T3 and a decrease in serum TSH. During this phase, there is no uptake of radioactive iodine in the thyroid. If thyrotoxicosis factitia is considered in the differential diagnosis, measurement of serum thyroglobulin levels is useful. During ingestion of levothyroxine, little or no thyroglobulin is present whereas serum thyroglobulin levels are elevated in silent thyroiditis. In 17 out of 71 patients with silent thyroiditis, moderate elevations of antithyroglobulin antibodies were present (213). Antimicrosomal antibodies were examined in 53 patients using the complement fixation test or by microsomal fluorescence. Using the former technique, 22 patients had positive antibodies, and by the latter 4 out of 7 were positive (213). In a small series of 7 patients with silent thyroiditis evaluated with a more sensitive radioimmuno assay (RIA) for human antithyroglobulin antibodies, all were positive (209). The white blood cell count is generally normal. In 53 episodes, 34 had elevated erythrocyte sedimentation rate (ESR), but it was greater than 40 mm/hr in only 8 (213). This contrasts with the typical marked elevation of the ESR in patients with subacute thyroiditis and helps to differentiate the two conditions. T4 and T3 reach subnormal levels in the hypothyroid range in 40% of patients (213). After the hypothyroid phase, patients gradually enter the euthyroid phase, heralded by an increase in thyroid hormone levels and resumption of thyroidal radioactive iodine uptake. The hypothyroid phase may last several months. In 26 episodes, patients became euthyroid after a mean period of 62 months after the onset of the hyperthyroid symptoms. TSH levels may increase during the recovery phase, and can remain elevated for many months. The delayed increase of TSH is due to its suppression during the thyrotoxic phase. Permanent hypothyroidism occurs in about 7% of patients with silent thyroiditis, but a subset of patients may ultimately become permanently hypothyroid. The echogenicity is decreased and a correlation between the decrease in the echo signal at the onset and nadir of the T3 level has been suggested (223) (Figure 13-5).
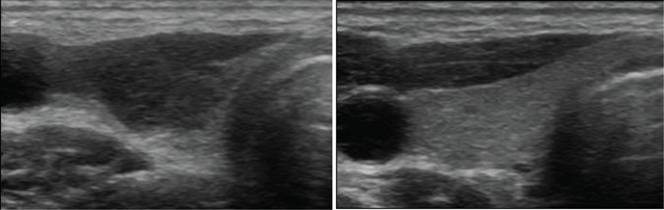
Treatment
As thyrotoxicosis is usually mild in silent thyroiditis, there is often no need for any treatment. In some patients, therapy with a beta-blocker can be considered during the thyrotoxic phase. In patients with more severe thyrotoxicosis, administration of NSAIDs and prednisone may be of benefit (224). After the thyrotoxic phase, many patients become temporarily hypothyroid and therapy with levothyroxine should be initiated in symptomatic patients. After a few months, levothyroxine therapy should be gradually withdrawn in order to assess whether the hypothyroidism is transient or permanent. Only a small proportion of patients remain permanently hypothyroid. Some patients, who initially recovered, may ultimately develop permanent thyroid failure (225). In a series of 54 patients, Nikolai et al. reported that about half of the patients developed permanent hypothyroidism (225). This is in contrast with subacute thyroiditis where permanent hypothyroidism is less common.
Pathogenesis
Although the disease was earlier considered to be a mild form of subacute (De Quervain's) thyroiditis, there is now convincing evidence that it is a lymphocytic thyroiditis (208, 209, 212-217, 222, 226). Many patients with silent thyroiditis have a personal or a family history of other autoimmune diseases, thereby indirectly supporting the concept that it is an autoimmune thyroiditis (227). There is no significant association with viral infections (211). There is a significant association with HLA genotype DR3. Postpartum thyroiditis (see below) is considered to be a form of silent thyroiditis occurring after delivery (228).
Pathology
On histological examination, follicles are disrupted and infiltrated by lymphocytes and plasma cells (211, 229). The infiltration is diffuse and/or focal, sometimes with the formation of lymphoid follicles. The follicular cells are heterogeneous in appearance. They can be cuboidal or columnar when stimulated by TSH. Some of the hypertrophic follicular cells have an oxyphilic cytoplasm (H�rthle or Ashkenazy cells). Thyroid tissue obtained during the hypothyroid or early recovery phase may show regenerating follicles with little colloid. In some patients, persistent mild lymphocytic thyroiditis is seen. Fibrosis is usually minimal, but can be extensive in some cases. Occasionally multinucleated giant cells, which are characteristic of subacute thyroiditis, are observed. The histological picture of postpartum thyroiditis is identical.
III. POSTPARTUM THYROIDITIS
Postpartum thyroiditis is considered to be a subform of silent (painless) thyroiditis (213). This condition is discussed in Chapter 14.
IV. HASHIMOTO'S THYROIDITIS
Occasionally, Hashimoto's thyroiditis is accompanied by mild symptoms of thyrotoxicosis, particularly in the early phases of the disease (230). This condition is discussed in Chapter 8.
THYROTOXICOSIS FACTITIA
Definition
Factitious thyrotoxicosis is due to the voluntary or involuntary intake of supraphysiological amounts of exogenous thyroid hormone (231). Most commonly, it is iatrogenic, either intentionally in order to suppress TSH in thyroid cancer patients or unintentionally in patients treated for primary hypothyroidism. In both instances, subclinical thyrotoxicosis is more common. The risk of atrial fibrilliation is increased in patients with long-standing suppression of TSH (232). Several cardiac parameters can be affected (233), but the severity of these effects is somewhat controversial (234). Suppressive doses of thyroid hormones can also affect bone mineral density (235), but this has not been confirmed in all studies (236). Non-iatrogenic thyrotoxicosis factitia can occur in patients of all ages with psychiatric illnesses (231, 237). In addition, some patients may take excessive amounts of thyroid hormones, sometimes prescribed by physicians, for weight loss, treatment of depression, or infertility (207). These patients often deny the intake of thyroid hormones or an excessive intake. In these instances, a heightened suspicion is needed in order to readily diagnose the disorder. Thyrotoxicosis induced by excessive thyroid hormone intake due to consumption of meat containing bovine thyroid tissue has been reported repeatedly. For example, two events of this so called "hamburger thyyrotoxicosis" have been documented in the United States (220, 221). Inclusion of the thyroid in neck muscle trimmings is now prohibited by US Department of Agriculture regulations. Accidental dosing with veterinary levothyroxine preparations has also been reported as a cause of thyrotoxicosis factitia (238).
Clinical presentation
Patients are clinically thyrotoxic, however they do not show signs of endocrine ophthalmopathy. In patients with a history of thyroid cancer, the history and the finding of a necklace scar readily provide an explanation. The thyroid may be small because of long-standing suppression of TSH.
Diagnosis
Serum TSH is suppressed, (free) T4 and T3 levels are variably elevated. The T4 and T3 levels depend on the type of ingested thyroid hormone preparation. Both T4 and T3 are high with excessive intake of levothyroxine, while only T3 is elevated with the intake of T3 preparations. With combination therapies, the T4 and T3 increases are variable depending on the relative amounts. Poisoning with T3 may be particularly severe (239), but even very high doses are often well tolerated, especially by children (240). When the diagnosis of thyrotoxicosis factitia is suspected, measurement of serum thyroglobulin levels is useful. During ingestion of levothyroxine, little or no thyroglobulin is present. In contrast, serum thyroglobulin levels are elevated in silent thyroiditis. Mariotti et al. performed thyroglobulin measurements in 6 women with thyrotoxicosis factitia (241). They used a sensitive thyroglobulin assay and excluded the presence of thyroglobulin antibodies that can potentially interfere with the assay. In all 6 women thyroglobulin was undetectable in the serum (241). However, thyroglobulin levels may not be reliable in patients who have anti-thyroglobulin antibodies. In these patients, measurement of fecal T4 can be used to distinguish endogenous and exogenous thyroid hormone excess (242). The thyroidal uptake of radioiodine or technetium are decreased. Doppler sonography shows absent thyroidal vascularity and low-normal peak systolic velocity (243). In contrast, these signs are increased in Graves' disease (243). Thus, factitious thyrotoxicosis is not difficult to differentiate from thyrotoxic Graves' disease, toxic adenoma or toxic multinodular goiter, or subacute thyroiditis. However, it may be difficult to readily distinguish silent thyroiditis from thyrotoxicosis factitia. In both situations, radioiodine uptake is very low or absent. In silent thyroiditis the serum thyroglobulin is, however, elevated. Suppressed radioactive uptake of the thyroid gland in combination with thyrotoxicosis may also exist in patients with hyperfunctioning metastases of well-differentiated thyroid carcinomas (76, 77). However, in these extremely rare patients, the thyroglobulin levels are elevated and radioactive iodine uptake will be detected in metastases by using whole body scanning.
Treatment
In most patients, adjustment or discontinuation of the thyroid hormone preparation is sufficient to normalize thyroid function tests. Patients with surreptitious intake of thyroid hormones for eating disorders or psychiatric illnesses can be difficult to treat and may need psychiatric consultation and assistance. In patients with severe intoxication, beta-blockers can be useful. Gastric lavage, induced emesis, activated charcoal, and, very rarely, plasmapheresis and exchange transfusion, can be considered in patients seen after acute ingestion of large amounts of thyroid hormone (231).
SUMMARY
Thyrotoxicosis can has a broad spectrum of etiologies (Table I). While it is most commonly caused by Graves' disease, it is of importance to recognize other etiologies in order to choose the most appropriate therapeutic option and long-term surveillance. Toxic adenomas are characterized by a single hyperactive nodule in the thyroid leading to clinical and biochemical thyrotoxicosis. Autonomous or toxic adenomas are most commonly caused by somatic gain-of-function mutations in the TSH receptor or the stimulatory Gs alpha subunit. A toxic adenoma is readily recognized on a thyroid scan. Toxic adenomas appear to be more common in countries with a low iodine intake. The possibility of developing thyrotoxicosis in a patient with a hot nodule with a diameter of 3 cm or larger is 20% in 6 years. This risk is substantially less in smaller nodules. Older patients with a hot nodule are more likely to become toxic as compared to younger patients. Definitive treatment consists in the administration of 131iodine, surgical removal of the nodule, or, less commonly used, percutaneous ethanol injection. The likelihood of malignancy in a toxic nodule is very low. In multinodular goiters, several nodules display an autonomous function. The pathogenesis is complex but may also include activating TSH receptor mutations. In addition to hyperthyroidism, some patients present with compressive signs. The diagnostic and therapeutic approach is in general similar to patients with a toxic adenoma, but may need cross-sectional imaging and pulmonary function tests in some patients. Therapeutically, surgery and radioiodine therapy are the most commonly used modalities. Well-differentiated thyroid carcinomas are only rarely associated with thyrotoxicosis. Treatment of patients with functioning thyroid carcinomas does not differ from the therapy of thyroid cancer patients without thyrotoxicosis, but appropriate control of the hyperthyroid state with antithyroid drugs and beta-blockers is important before submitting a patient to thyroid surgery or 131iodine therapy. Familial and sporadic forms of non-autoimmune hyperthyroidism are uncommon. They are caused by inherited or de novo germline gain-of-function mutations in the TSH receptor. Inappropriate TSH secretion by a TSH-secreting pituitary tumor is a rare cause of hyperthyroidism. Transsphenoidal surgery, in combination with radiotherapy and somatostatin analogues in some patients, are the therapies of choice. During pregnancy, transient gestational thyrotoxicosis may be due to stimulation of the TSH receptor by high levels of hCG. In a single instance, a mutation in the TSH receptor conferring hypersensitivity to hCG has been reported. Hydatiform moles or a choriocarcinomas can lead to high hCG levels and thyrotoxicosis. Hydatiform moles are treated by suction. Choriocarcinomas can now be treated successfully in most patients with chemotherapy. Struma ovarii, thyroid tissue in a ovarian teratoma, rarely causes hyperthyroidism. Most patients with struma ovarii are clinically and biochemically euthyroid. Treatment consists of surgical removal of the teratoma. Administration of moderate or high doses of iodine may induce thyrotoxicosis in patients with or without apparent pre-existing thyroid disease. There are numerous sources of iodine, for example drugs, contrast agents, disinfectants, and food components. A notorious iodine-containing agent is the anti-arrhythmic drug amiodarone, which may induce thyrotoxicosis because of its high iodine content and/or a drug-induced thyroiditis. Any form of thyroiditis can be associated with a thyrotoxic phase because the disruption of thyroid follicles can result in an increased release of stored iodothyronines. The thyrotoxic phase may be followed by transient or permanent hypothyroidism. All forms of thyroiditis can be associated with a thyrotoxic phase because the disruption of thyroid follicles can result in an increased release of stored iodothyronines. The thyrotoxic phase may be followed by transient or permanent hypothyroidism. The uptake of radioiodine is very low or absent in the thyrotoxic phase and serum thyroglobulin levels are high. Clinical thyrotoxicosis is often mild and treatment with beta-blocking agents is often sufficient. Although the majority of patients recover, a substantial subset of patients develops hypothyroidism in later years. Therefore, regular assessment of thyroid function is necessary. Thyrotoxicosis factitia, the excessive intake of exogenous thyroid hormones, can be iatrogenic, or due to voluntary or involuntary intake of thyroid hormones. The uptake of radioiodine is low and thyroglobulin levels are also very low or undetectable. The thyroid may be small. The therapy consists in appropriate dose adjustment or discontinuation of exogenous thyroid hormone.
ACKNOWLEDGMENT
This chapter is, in part, based on the previous version written by Dr. Georg Hennemann. These contributions are gratefully acknowledged.
- 1.
- Braverman L, Utiger R: Introduction to thyrotoxicosis. In: Braverman L, Utiger R eds. The Thyroid. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 453-455, 2005.
- 2.
- Reinwein D, Benker G, Konig MP, et al.: The different types of hyperthyroidism in Europe. Results of a prospective survey of 924 patients. J Endocrinol Invest 11:193-200, 1988. [PubMed: 3372959]
- 3.
- Hamburger JI: Evolution of toxicity in solitary nontoxic autonomously functioning thyroid nodules. J Clin Endocrinol Metab 50:1089-1093, 1980. [PubMed: 7372787]
- 4.
- Bransom CJ, Talbot CH, Henry L, et al.: Solitary toxic adenoma of the thyroid gland. Br J Surg 66:592-595, 1979. [PubMed: 486923]
- 5.
- Horst W, Rosler H, Schneider C, et al.: 306 cases of toxic adenoma: clinical aspects, findings in radioiodine diagnostics, radiochromatography and histology; results of 131-I and surgical treatment. J Nucl Med 8:515-528, 1967. [PubMed: 4166326]
- 6.
- Orgiazzi J, Mornex R: Hyperthyroidism. In: Greer M ed. The thyroid gland. New York: Raven Press 442, 1990.
- 7.
- Berglund J, Ericsson UB, Hallengren B: Increased incidence of thyrotoxicosis in Malmo during the years 1988-1990 as compared to the years 1970-1974. J Intern Med 239:57-62, 1996. [PubMed: 8551201]
- 8.
- Kopp P, Muirhead S, Jourdain N, et al.: Congenital hyperthyroidism caused by a solitary toxic adenoma harboring a novel somatic mutation (serine281-->isoleucine) in the extracellular domain of the thyrotropin receptor. J Clin Invest 100:1634-1639, 1997. [PMC free article: PMC508345] [PubMed: 9294132]
- 9.
- Emrich D, Erlenmaier U, Pohl M, et al.: Determination of the autonomously functioning volume of the thyroid. Eur J Nucl Med 20:410-414, 1993. [PubMed: 8390935]
- 10.
- Baloch Z, Carayon P, Conte-Devolx B, et al.: Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid 13:3-126, 2003. [PubMed: 12625976]
- 11.
- Hegedus L: Clinical practice. The thyroid nodule. N Engl J Med 351:1764-1771, 2004. [PubMed: 15496625]
- 12.
- Thomas CG, Jr., Croom RD, 3rd: Current management of the patient with autonomously functioning nodular goiter. Surg Clin North Am 67:315-328, 1987. [PubMed: 3551148]
- 13.
- B�hre M, Hilgers R, Lindemann C, et al.: Thyroid autonomy: sensitive detection in vivo and estimation of its functional relevance using quantified high-resolution scintigraphy. Acta Endocrinol (Copenh) 117:145-153, 1988. [PubMed: 2837884]
- 14.
- Sandler MP, Fellmeth B, Salhany KE, et al.: Thyroid carcinoma masquerading as a solitary benign hyperfunctioning nodule. Clin Nucl Med 13:410-415, 1988. [PubMed: 3402143]
- 15.
- Cibas ES, Ali SZ: The Bethesda System For Reporting Thyroid Cytopathology. Am J Clin Pathol 132:658-665, 2009. [PubMed: 19846805]
- 16.
- Ferrari C, Reschini E, Paracchi A: Treatment of the autonomous thyroid nodule: a review. Eur J Endocrinol 135:383-390, 1996. [PubMed: 8921817]
- 17.
- Eyre-Brook IA, Talbot CH: The treatment of autonomous functioning thyroid nodules. Br J Surg 69:577-579, 1982. [PubMed: 7127035]
- 18.
- Goldstein R, Hart IR: Follow-up of solitary autonomous thyroid nodules treated with 131I. N Engl J Med 309:1473-1476, 1983. [PubMed: 6646172]
- 19.
- Mariotti S, Martino E, Francesconi M, et al.: Serum thyroid autoantibodies as a risk factor for development of hypothyroidism after radioactive iodine therapy for single thyroid 'hot' nodule. Acta Endocrinol (Copenh) 113:500-507, 1986. [PubMed: 3788420]
- 20.
- Ratcliffe GE, Cooke S, Fogelman I, et al.: Radioiodine treatment of solitary functioning thyroid nodules. Br J Radiol 59:385-387, 1986. [PubMed: 3697616]
- 21.
- Ross DS, Ridgway EC, Daniels GH: Successful treatment of solitary toxic thyroid nodules with relatively low-dose iodine-131, with low prevalence of hypothyroidism. Ann Intern Med 101:488-490, 1984. [PubMed: 6476634]
- 22.
- Bolusani H, Okosieme OE, Velagapudi M, et al.: Determinants of long-term outcome after radioiodine therapy for solitary autonomous thyroid nodules. Endocr Pract 14:543-549, 2008. [PubMed: 18753095]
- 23.
- Hovens GC, Heemstra KA, Buiting AM, et al.: Induction of stimulating thyrotropin receptor antibodies after radioiodine therapy for toxic multinodular goitre and Graves' disease measured with a novel bioassay. Nucl Med Commun 28:123-127, 2007. [PubMed: 17198353]
- 24.
- Nygaard B, Knudsen JH, Hegedus L, et al.: Thyrotropin receptor antibodies and Graves' disease, a side-effect of 131I treatment in patients with nontoxic goiter. J Clin Endocrinol Metab 82:2926-2930, 1997. [PubMed: 9284721]
- 25.
- Lippi F, Ferrari C, Manetti L, et al.: Treatment of solitary autonomous thyroid nodules by percutaneous ethanol injection: results of an Italian multicenter study. The Multicenter Study Group. J Clin Endocrinol Metab 81:3261-3264, 1996. [PubMed: 8784080]
- 26.
- Monzani F, Caraccio N, Goletti O, et al.: Five-year follow-up of percutaneous ethanol injection for the treatment of hyperfunctioning thyroid nodules: a study of 117 patients. Clin Endocrinol (Oxf) 46:9-15, 1997. [PubMed: 9059552]
- 27.
- Zingrillo M, Torlontano M, Ghiggi MR, et al.: Radioiodine and percutaneous ethanol injection in the treatment of large toxic thyroid nodule: a long-term study. Thyroid 10:985-989, 2000. [PubMed: 11128727]
- 28.
- Del Prete S, Russo D, Caraglia M, et al.: Percutaneous ethanol injection of autonomous thyroid nodules with a volume larger than 40 ml: three years of follow-up. Clin Radiol 56:895-901, 2001. [PubMed: 11603892]
- 29.
- Monzani F, Caraccio N, Basolo F, et al.: Surgical and pathological changes after percutaneous ethanol injection therapy of thyroid nodules. Thyroid 10:1087-1092, 2000. [PubMed: 11201854]
- 30.
- Pacella CM, Bizzarri G, Spiezia S, et al.: Thyroid tissue: US-guided percutaneous laser thermal ablation. Radiology 232:272-280, 2004. [PubMed: 15155898]
- 31.
- Papini E, Bizzarri G, Pacella CM: Percutaneous laser ablation of benign and malignant thyroid nodules. Curr Opin Endocrinol Diabetes Obes 15:434-439, 2008. [PubMed: 18769216]
- 32.
- Deandrea M, Limone P, Basso E, et al.: US-guided percutaneous radiofrequency thermal ablation for the treatment of solid benign hyperfunctioning or compressive thyroid nodules. Ultrasound Med Biol 34:784-791, 2008. [PubMed: 18207307]
- 33.
- Dumont JE, Lamy F, Roger P, et al.: Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev 72:667-697, 1992. [PubMed: 1320763]
- 34.
- Lyons J, Landis CA, Harsh G, et al.: Two G protein oncogenes in human endocrine tumors. Science 249:655-659, 1990. [PubMed: 2116665]
- 35.
- O'Sullivan C, Barton CM, Staddon SL, et al.: Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol Carcinog 4:345-349, 1991. [PubMed: 1910478]
- 36.
- Suarez HG, du Villard JA, Caillou B, et al.: gsp mutations in human thyroid tumours. Oncogene 6:677-679, 1991. [PubMed: 1903197]
- 37.
- Spada A, Vallar L, Faglia G: G protein oncogenes in pituitary tumors. Trends Endocrinol Metab 3:355-360, 1992. [PubMed: 18407121]
- 38.
- Weinstein LS, Shenker A, Gejman PV, et al.: Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325:1688-1695, 1991. [PubMed: 1944469]
- 39.
- Kjelsberg MA, Cotecchia S, Ostrowski J, et al.: Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J Biol Chem 267:1430-1433, 1992. [PubMed: 1346134]
- 40.
- Parma J, Duprez L, Van Sande J, et al.: Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651, 1993. [PubMed: 8413627]
- 41.
- Van Sande J, Parma J, Tonacchera M, et al.: Somatic and germline mutations of the TSH receptor gene in thyroid diseases. J Clin Endocrinol Metab 80:2577-2585, 1995. [PubMed: 7673398]
- 42.
- Paschke R, Ludgate M: The thyrotropin receptor in thyroid diseases. N Engl J Med 337:1675-1681, 1997. [PubMed: 9385128]
- 43.
- Parma J, Van Sande J, Swillens S, et al.: Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3',5'-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol 9:725-733, 1995. [PubMed: 8592518]
- 44.
- Kopp P: The TSH receptor and its role in thyroid disease. Cell Mol Life Sci 58:1301-1322, 2001. [PubMed: 11577986]
- 45.
- Takeshita A, Nagayama Y, Yokoyama N, et al.: Rarity of oncogenic mutations in the thyrotropin receptor of autonomously functioning thyroid nodules in Japan. J Clin Endocrinol Metab 80:2607-2611, 1995. [PubMed: 7673402]
- 46.
- Parma J, Duprez L, Van Sande J, et al.: Diversity and prevalence of somatic mutations in the thyrotropin receptor and Gs alpha genes as a cause of toxic thyroid adenomas. J Clin Endocrinol Metab 82:2695-2701, 1997. [PubMed: 9253356]
- 47.
- Tonacchera M, Cetani F, Parma J, et al.: Oncogenic mutations in thyroid adenoma: methodological criteria. Eur J Endocrinol 135:444-446, 1996. [PubMed: 8921827]
- 48.
- Holzapfel HP, Bergner B, Wonerow P, et al.: Expression of G(alpha)(s) proteins and TSH receptor signalling in hyperfunctioning thyroid nodules with TSH receptor mutations. Eur J Endocrinol 147:109-116, 2002. [PubMed: 12088927]
- 49.
- Trulzsch B, Krohn K, Wonerow P, et al.: Detection of thyroid-stimulating hormone receptor and Gsalpha mutations: in 75 toxic thyroid nodules by denaturing gradient gel electrophoresis. J Mol Med 78:684-691, 2001. [PubMed: 11434721]
- 50.
- Paschke R, Tonacchera M, Van Sande J, et al.: Identification and functional characterization of two new somatic mutations causing constitutive activation of the thyrotropin receptor in hyperfunctioning autonomous adenomas of the thyroid. J Clin Endocrinol Metab 79:1785-1789, 1994. [PubMed: 7989485]
- 51.
- Duprez L, Parma J, Van Sande J, et al.: Germline mutations in the thyrotropin receptor gene cause non-autoimmune autosomal dominant hyperthyroidism. Nat Genet 7:396-401, 1994. [PubMed: 7920658]
- 52.
- Duprez L, Parma J, Costagliola S, et al.: Constitutive activation of the TSH receptor by spontaneous mutations affecting the N-terminal extracellular domain. FEBS Lett 409:469-474, 1997. [PubMed: 9224711]
- 53.
- Zhang ML, Sugawa H, Kosugi S, et al.: Constitutive activation of the thyrotropin receptor by deletion of a portion of the extracellular domain. Biochem Biophys Res Commun 211:205-210, 1995. [PubMed: 7779086]
- 54.
- Van Sande J, Massart C, Costagliola S, et al.: Specific activation of the thyrotropin receptor by trypsin. Mol Cell Endocrinol 119:161-168, 1996. [PubMed: 8807635]
- 55.
- Krohn K, Paschke R: Somatic mutations in thyroid nodular disease. Mol Genet Metab 75:202-208, 2002. [PubMed: 11914031]
- 56.
- Derwahl M, Studer H: Nodular goiter and goiter nodules: Where iodine deficiency falls short of explaining the facts. Exp Clin Endocrinol Diabetes 109:250-260, 2001. [PubMed: 11507648]
- 57.
- Hedinger C, Williams ED, Sobin LH: The WHO histological classification of thyroid tumors: a commentary on the second edition. Cancer 63:908-911, 1989. [PubMed: 2914297]
- 58.
- Deleu S, Allory Y, Radulescu A, et al.: Characterization of autonomous thyroid adenoma: metabolism, gene expression, and pathology. Thyroid 10:131-140, 2000. [PubMed: 10718549]
- 59.
- Krohn K, Emmrich P, Ott N, et al.: Increased thyroid epithelial cell proliferation in toxic thyroid nodules. Thyroid 9:241-246, 1999. [PubMed: 10211599]
- 60.
- Becker FO, Economou PG, Schwartz TB: The occurrence of carcinoma in "hot" thyroid nodules. Report of two cases. Ann Intern Med 58:877-882, 1963. [PubMed: 13970110]
- 61.
- Fujimoto Y, Oka A, Nagataki S: Occurrence of papillary carcinoma in hyperfunctioning thyroid nodule. Report of a case. Endocrinol Jpn 19:371-374, 1972. [PubMed: 4678652]
- 62.
- Smith M, McHenry C, Jarosz H, et al.: Carcinoma of the thyroid in patients with autonomous nodules. Am Surg 54:448-449, 1988. [PubMed: 3389595]
- 63.
- Hamburger JI: Solitary autonomously functioning thyroid lesions. Diagnosis, clinical features and pathogenetic considerations. Am J Med 58:740-748, 1975. [PubMed: 1138537]
- 64.
- Nikiforov Y, Thompson L, Biddinger P: Diagnostic Surgical Pathology and Molecular Genetics of the Thyroid Philadelphia: Lippincott Williams & Wilkins, 2010.
- 65.
- Studer H, Derwahl M: Mechanisms of nonneoplastic endocrine hyperplasia--a changing concept: a review focused on the thyroid gland. Endocr Rev 16:411-426, 1995. [PubMed: 8521787]
- 66.
- Duprez L, Hermans J, Van Sande J, et al.: Two autonomous nodules of a patient with multinodular goiter harbor different activating mutations of the thyrotropin receptor gene. J Clin Endocrinol Metab 82:306-308, 1997. [PubMed: 8989278]
- 67.
- Holzapfel HP, Fuhrer D, Wonerow P, et al.: Identification of constitutively activating somatic thyrotropin receptor mutations in a subset of toxic multinodular goiters. J Clin Endocrinol Metab 82:4229-4233, 1997. [PubMed: 9398745]
- 68.
- Tonacchera M, Chiovato L, Pinchera A, et al.: Hyperfunctioning thyroid nodules in toxic multinodular goiter share activating thyrotropin receptor mutations with solitary toxic adenoma. J Clin Endocrinol Metab 83:492-498, 1998. [PubMed: 9467563]
- 69.
- Kopp P, Kimura ET, Aeschimann S, et al.: Polyclonal and monoclonal thyroid nodules coexist within human multinodular goiters. J Clin Endocrinol Metab 79:134-139, 1994. [PubMed: 7517946]
- 70.
- Tonacchera M, Agretti P, Chiovato L, et al.: Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 85:2270-2274, 2000. [PubMed: 10852462]
- 71.
- Gabriel EM, Bergert ER, Grant CS, et al.: Germline polymorphism of codon 727 of human thyroid-stimulating hormone receptor is associated with toxic multinodular goiter. J Clin Endocrinol Metab 84:3328-3335, 1999. [PubMed: 10487707]
- 72.
- Nogueira CR, Kopp P, Arseven OK, et al.: Thyrotropin receptor mutations in hyperfunctioning thyroid adenomas from Brazil. Thyroid 9:1063-1068, 1999. [PubMed: 10595453]
- 73.
- Muhlberg T, Herrmann K, Joba W, et al.: Lack of association of nonautoimmune hyperfunctioning thyroid disorders and a germline polymorphism of codon 727 of the human thyrotropin receptor in a European Caucasian population. J Clin Endocrinol Metab 85:2640-2643, 2000. [PubMed: 10946859]
- 74.
- Krohn K, Wohlgemuth S, Gerber H, et al.: Hot microscopic areas of iodine-deficient euthyroid goitres contain constitutively activating TSH receptor mutations. J Pathol 192:37-42, 2000. [PubMed: 10951398]
- 75.
- Kopp P, Wilkes B, Gu W, et al.: Absence of mutations in the TSHR receptor and the Gsa subunit in nodules and adenomas of multinodular goiters and identification of a new mutation (tyrosine601>asparagine) in the fifth transmembrane domain in a toxic adenoma. . Thyroid, 69th meeting of the American Thyroid Association, San Diego 6:S-9, Abstract P17, 1996.
- 76.
- Ehrenheim C, Heintz P, Schober O, et al.: Jodinduzierte T3-Hyperthyreose beim metastasierenden follikularen Schilddrusenkarzinom. Nuklearmedizin 25:201-204, 1986. [PubMed: 3797260]
- 77.
- Salvatori M, Saletnich I, Rufini V, et al.: Severe thyrotoxicosis due to functioning pulmonary metastases of well-differentiated thyroid cancer. J Nucl Med 39:1202-1207, 1998. [PubMed: 9669394]
- 78.
- Nakashima T, Inoue K, Shiro-ozu A, et al.: Predominant T3 synthesis in the metastatic thyroid carcinoma in a patient with T3-toxicosis. Metabolism 30:327-330, 1981. [PubMed: 7207204]
- 79.
- Paul SJ, Sisson JC: Thyrotoxicosis caused by thyroid cancer. Endocrinol Metab Clin North Am 19:593-612, 1990. [PubMed: 2261908]
- 80.
- Russo D, Tumino S, Arturi F, et al.: Detection of an activating mutation of the thyrotropin receptor in a case of an autonomously hyperfunctioning thyroid insular carcinoma. J Clin Endocrinol Metab 82:735-738, 1997. [PubMed: 9062474]
- 81.
- Cerletty JM, Listwan WJ: Hyperthyroidism due to functioning metastatic thyroid carcinoma. Precipitation of thyroid storm with therapeutic radioactive iodine. JAMA 242:269-270, 1979. [PubMed: 448918]
- 82.
- Matsuo K, Friedman E, Gejman PV, et al.: The thyrotropin receptor (TSH-R) is not an oncogene for thyroid tumors: structural studies of the TSH-R and the alpha-subunit of Gs in human thyroid neoplasms. J Clin Endocrinol Metab 76:1446-1451, 1993. [PubMed: 8501149]
- 83.
- Russo D, Arturi F, Schlumberger M, et al.: Activating mutations of the TSH receptor in differentiated thyroid carcinomas. Oncogene 11:1907-1911, 1995. [PubMed: 7478621]
- 84.
- Spambalg D, Sharifi N, Elisei R, et al.: Structural studies of the thyrotropin receptor and Gs alpha in human thyroid cancers: low prevalence of mutations predicts infrequent involvement in malignant transformation. J Clin Endocrinol Metab 81:3898-3901, 1996. [PubMed: 8923835]
- 85.
- Russo D, Wong MG, Costante G, et al.: A Val 677 activating mutation of the thyrotropin receptor in a Hurthle cell thyroid carcinoma associated with thyrotoxicosis. Thyroid 9:13-17, 1999. [PubMed: 10037070]
- 86.
- Camacho P, Gordon D, Chiefari E, et al.: A Phe 486 thyrotropin receptor mutation in an autonomously functioning follicular carcinoma that was causing hyperthyroidism. Thyroid 10:1009-1012, 2000. [PubMed: 11128715]
- 87.
- Mircescu H, Parma J, Huot C, et al.: Hyperfunctioning malignant thyroid nodule in an 11-year-old girl: pathologic and molecular studies. J Pediatr 137:585-587, 2000. [PubMed: 11035845]
- 88.
- de Roux N, Polak M, Couet J, et al.: A neomutation of the thyroid-stimulating hormone receptor in a severe neonatal hyperthyroidism. J Clin Endocrinol Metab 81:2023-2026, 1996. [PubMed: 8964822]
- 89.
- Lavard L, Sehested A, Brock Jacobsen B, et al.: Long-term follow-Up of an infant with thyrotoxicosis due to germline mutation of the TSH receptor gene (Met453Thr). Horm Res 51:43-46, 1999. [PubMed: 10095169]
- 90.
- Fournes B, Monier R, Michiels F, et al.: Oncogenic potential of a mutant human thyrotropin receptor expressed in FRTL-5 cells. Oncogene 16:985-990, 1998. [PubMed: 9519872]
- 91.
- Grayzel EF, Bennett B: Graves' disease, follicular thyroid carcinoma and functioning pulmonary metastases. Cancer 43:1885-1887, 1979. [PubMed: 582160]
- 92.
- Kasagi K, Takeuchi R, Miyamoto S, et al.: Metastatic thyroid cancer presenting as thyrotoxicosis: report of three cases. Clin Endocrinol (Oxf) 40:429-434, 1994. [PubMed: 8187309]
- 93.
- Steffensen FH, Aunsholt NA: Hyperthyroidism associated with metastatic thyroid carcinoma. Clin Endocrinol (Oxf) 41:685-687, 1994. [PubMed: 7828360]
- 94.
- Mazzaferri EL: Thyroid cancer and Graves' disease. J Clin Endocrinol Metab 70:826-829, 1990. [PubMed: 2180977]
- 95.
- Belfiore A, Garofalo MR, Giuffrida D, et al.: Increased aggressiveness of thyroid cancer in patients with Graves' disease. J Clin Endocrinol Metab 70:830-835, 1990. [PubMed: 2180978]
- 96.
- Hales IB, McElduff A, Crummer P, et al.: Does Graves' disease or thyrotoxicosis affect the prognosis of thyroid cancer. J Clin Endocrinol Metab 75:886-889, 1992. [PubMed: 1517381]
- 97.
- Thomas JS, Leclere J, Hartemann P, et al.: Familial hyperthyroidism without evidence of autoimmunity. Acta Endocrinol (Copenh) 100:512-518, 1982. [PubMed: 7124278]
- 98.
- Gozu HI, Lublinghoff J, Bircan R, et al.: Genetics and phenomics of inherited and sporadic non-autoimmune hyperthyroidism. Mol Cell Endocrinol 322:125-134, 2010. [PubMed: 20138963]
- 99.
- Horton G: Hyperthyroidie h�reditaire par hyperactivit� diffuse non-autoimmune de la thyroide avec autonomie de fonction et de croissance. In. Lausanne, Switzerland: University of Lausanne1987.
- 100.
- Leclere J, Bene MC, Aubert V, et al.: Clinical consequences of activating germline mutations of TSH receptor, the concept of toxic hyperplasia. Horm Res 47:158-162, 1997. [PubMed: 9167947]
- 101.
- Ramsay I, Kaur S, Krassas G: Thyrotoxicosis in pregnancy: results of treatment by antithyroid drugs combined with T4. Clin Endocrinol (Oxf) 18:73-85, 1983. [PubMed: 6682725]
- 102.
- Zakarija M, McKenzie JM: Pregnancy-associated changes in the thyroid-stimulating antibody of Graves' disease and the relationship to neonatal hyperthyroidism. J Clin Endocrinol Metab 57:1036-1040, 1983. [PubMed: 6137493]
- 103.
- Zakarija M, McKenzie JM, Hoffman WH: Prediction and therapy of intrauterine and late-onset neonatal hyperthyroidism. J Clin Endocrinol Metab 62:368-371, 1986. [PubMed: 2867106]
- 104.
- Kopp P, van Sande J, Parma J, et al.: Brief report: congenital hyperthyroidism caused by a mutation in the thyrotropin-receptor gene. N Engl J Med 332:150-154, 1995. [PubMed: 7800007]
- 105.
- Kopp P, Jameson JL, Roe TF: Congenital nonautoimmune hyperthyroidism in a nonidentical twin caused by a sporadic germline mutation in the thyrotropin receptor gene. Thyroid 7:765-770, 1997. [PubMed: 9349581]
- 106.
- Holzapfel HP, Wonerow P, von Petrykowski W, et al.: Sporadic congenital hyperthyroidism due to a spontaneous germline mutation in the thyrotropin receptor gene. J Clin Endocrinol Metab 82:3879-3884, 1997. [PubMed: 9360555]
- 107.
- Hollingsworth D: Neonatal hyperthyroidism. In: Delange F, Fisher D, Malvaux P eds. Pediatric Thyroidology. Basel: Karger; 210-222, 1985.
- 108.
- Beck-Peccoz P, Persani L, Mannavola D, et al.: Pituitary tumours: TSH-secreting adenomas. Best Pract Res Clin Endocrinol Metab 23:597-606, 2009. [PubMed: 19945025]
- 109.
- Beck-Peccoz P, Brucker-Davis F, Persani L, et al.: Thyrotropin-secreting pituitary tumors. Endocr Rev 17:610-638, 1996. [PubMed: 8969971]
- 110.
- Nakayama Y, Jinguji S, Kumakura SI, et al.: Thyroid-stimulating hormone (thyrotropin)-secretion pituitary adenoma in an 8-year-old boy: case report. Pituitary2010. [PubMed: 21113740]
- 111.
- McDermott MT, Ridgway EC: Central hyperthyroidism. Endocrinol Metab Clin North Am 27:187-203, 1998. [PubMed: 9534036]
- 112.
- Socin HV, Chanson P, Delemer B, et al.: The changing spectrum of TSH-secreting pituitary adenomas: diagnosis and management in 43 patients. Eur J Endocrinol 148:433-442, 2003. [PubMed: 12656664]
- 113.
- Sergi I, Medri G, Papandreou MJ, et al.: Polymorphism of thyrotropin and alpha subunit in human pituitary adenomas. J Endocrinol Invest 16:45-55, 1993. [PubMed: 8445156]
- 114.
- Roelfsema F, Pereira AM, Keenan DM, et al.: Thyrotropin secretion by thyrotropinomas is characterized by increased pulse frequency, delayed diurnal rhythm, enhanced basal secretion, spikiness, and disorderliness. J Clin Endocrinol Metab 93:4052-4057, 2008. [PubMed: 18682501]
- 115.
- Brucker-Davis F, Oldfield EH, Skarulis MC, et al.: Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab 84:476-486, 1999. [PubMed: 10022404]
- 116.
- Kienitz T, Quinkler M, Strasburger CJ, et al.: Long-term management in five cases of TSH-secreting pituitary adenomas: a single center study and review of the literature. Eur J Endocrinol 157:39-46, 2007. [PubMed: 17609400]
- 117.
- Gatto F, Barbieri F, Castelletti L, et al.: In vivo and in vitro response to octreotide LAR in a TSH-secreting adenoma: characterization of somatostatin receptor expression and role of subtype 5. Pituitary2010. [PubMed: 21086053]
- 118.
- Refetoff S, Weiss RE, Usala SJ: The syndromes of resistance to thyroid hormone. Endocr Rev 14:348-399, 1993. [PubMed: 8319599]
- 119.
- Refetoff S, Dumitrescu AM: Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab 21:277-305, 2007. [PubMed: 17574009]
- 120.
- Flamant F, Samarut J: Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab 14:85-90, 2003. [PubMed: 12591179]
- 121.
- Glinoer D, De Nayer P, Robyn C, et al.: Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 16:881-888, 1993. [PubMed: 7511622]
- 122.
- Yoshimura M, Hershman JM: Thyrotropic action of human chorionic gonadotropin. Thyroid 5:425-434, 1995. [PubMed: 8563483]
- 123.
- Glinoer D, Lemone M: Goiter and pregnancy: a new insight into an old problem. Thyroid 2:65-70, 1992. [PubMed: 1525569]
- 124.
- Glinoer D: Thyroid hyperfunction during pregnancy. Thyroid 8:859-864, 1998. [PubMed: 9777758]
- 125.
- Trzepacz PT, Klein I, Roberts M, et al.: Graves' disease: an analysis of thyroid hormone levels and hyperthyroid signs and symptoms. Am J Med 87:558-561, 1989. [PubMed: 2816972]
- 126.
- Goodwin TM, Montoro M, Mestman JH, et al.: The role of chorionic gonadotropin in transient hyperthyroidism of hyperemesis gravidarum. J Clin Endocrinol Metab 75:1333-1337, 1992. [PubMed: 1430095]
- 127.
- Glinoer D: The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endocr Rev 18:404-433, 1997. [PubMed: 9183570]
- 128.
- Roti E, Gardini E, Minelli R, et al.: Thyroid function evaluation by different commercially available free thyroid hormone measurement kits in term pregnant women and their newborns. J Endocrinol Invest 14:1-9, 1991. [PubMed: 2045620]
- 129.
- Cooper DS, Rivkees SA: Putting propylthiouracil in perspective. J Clin Endocrinol Metab 94:1881-1882, 2009. [PubMed: 19401361]
- 130.
- Bahn RS, Burch HS, Cooper DS, et al.: The Role of Propylthiouracil in the Management of Graves' Disease in Adults: report of a meeting jointly sponsored by the American Thyroid Association and the Food and Drug Administration. Thyroid 19:673-674, 2009. [PubMed: 19583480]
- 131.
- Mandel SJ, Cooper DS: The use of antithyroid drugs in pregnancy and lactation. J Clin Endocrinol Metab 86:2354-2359, 2001. [PubMed: 11397822]
- 132.
- Hershman JM: Physiological and pathological aspects of the effect of human chorionic gonadotropin on the thyroid. Best Pract Res Clin Endocrinol Metab 18:249-265, 2004. [PubMed: 15157839]
- 133.
- Burrow GN: Thyroid function and hyperfunction during gestation. Endocr Rev 14:194-202, 1993. [PubMed: 8325252]
- 134.
- Glinoer D, de Nayer P, Bourdoux P, et al.: Regulation of maternal thyroid during pregnancy. J Clin Endocrinol Metab 71:276-287, 1990. [PubMed: 2116437]
- 135.
- Grun JP, Meuris S, De Nayer P, et al.: The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 46:719-725, 1997. [PubMed: 9274703]
- 136.
- Carayon P, Lefort G, Nisula B: Interaction of human chorionic gonadotropin and human luteinizing hormone with human thyroid membranes. Endocrinology 106:1907-1916, 1980. [PubMed: 6245856]
- 137.
- Hoermann R, Amir SM, Ingbar SH: Evidence that partially desialylated variants of human chorionic gonadotropin (hCG) are the factors in crude hCG that inhibit the response to thyrotropin in human thyroid membranes. Endocrinology 123:1535-1543, 1988. [PubMed: 3402396]
- 138.
- Davies TF, Platzer M: hCG-induced TSH receptor activation and growth acceleration in FRTL-5 thyroid cells. Endocrinology 118:2149-2151, 1986. [PubMed: 3009152]
- 139.
- Hershman JM, Lee HY, Sugawara M, et al.: Human chorionic gonadotropin stimulates iodide uptake, adenylate cyclase, and deoxyribonucleic acid synthesis in cultured rat thyroid cells. J Clin Endocrinol Metab 67:74-79, 1988. [PubMed: 3379138]
- 140.
- Rodien P, Bremont C, Sanson ML, et al.: Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. N Engl J Med 339:1823-1826, 1998. [PubMed: 9854118]
- 141.
- Seckl MJ, Sebire NJ, Berkowitz RS: Gestational trophoblastic disease. Lancet 376:717-729, 2010. [PubMed: 20673583]
- 142.
- Hershman J: Trophoblastic tumors. In: Braverman L, Utiger R eds. The Thyroid 9th ed ed. Philadelphia: Lippincott-Raven; 519-523, 2005.
- 143.
- Hershman JM, Higgins HP: Hydatidiform mole--a cause of clinical hyperthyroidism. Report of two cases with evidence that the molar tissue secreted a thyroid stimulator. N Engl J Med 284:573-577, 1971. [PubMed: 5100890]
- 144.
- Gleason PE, Elliott DS, Zimmerman D, et al.: Metastatic testicular choriocarcinoma and secondary hyperthyroidism: case report and review of the literature. J Urol 151:1063-1064, 1994. [PubMed: 8126794]
- 145.
- Nisula BC, Taliadouros GS: Thyroid function in gestational trophoblastic neoplasia: evidence that the thyrotropic activity of chorionic gonadotropin mediates the thyrotoxicosis of choriocarcinoma. Am J Obstet Gynecol 138:77-85, 1980. [PubMed: 6251724]
- 146.
- Anderson NR, Lokich JJ, McDermott WV, Jr., et al.: Gestational choriocarcinoma and thyrotoxicosis. Cancer 44:304-306, 1979. [PubMed: 455255]
- 147.
- Orgiazzi J, Rousset B, Cosentino C, et al.: Plasma thyrotropic activity in a man with choriocarcinoma. J Clin Endocrinol Metab 39:653-657, 1974. [PubMed: 4472238]
- 148.
- Noal S, Joly F, Leblanc E: [Management of gestational trophoblastic disease]. Gynecol Obstet Fertil 38:193-198, 2010. [PubMed: 20189434]
- 149.
- Schmid P, Nagai Y, Agarwal R, et al.: Prognostic markers and long-term outcome of placental-site trophoblastic tumours: a retrospective observational study. Lancet 374:48-55, 2009. [PubMed: 19552948]
- 150.
- Roth LM, Talerman A: The enigma of struma ovarii. Pathology 39:139-146, 2007. [PubMed: 17365830]
- 151.
- Dardik RB, Dardik M, Westra W, et al.: Malignant struma ovarii: two case reports and a review of the literature. Gynecol Oncol 73:447-451, 1999. [PubMed: 10366477]
- 152.
- Yassa L, Sadow P, Marqusee E: Malignant struma ovarii. Nat Clin Pract Endocrinol Metab 4:469-472, 2008. [PubMed: 18560398]
- 153.
- Dunzendorfer T, deLas Morenas A, Kalir T, et al.: Struma ovarii and hyperthyroidism. Thyroid 9:499-502, 1999. [PubMed: 10365682]
- 154.
- Paladini D, Vassallo M, Sglavo G, et al.: Struma ovarii associated with hyperthyroidism, elevated CA 125 and pseudo-Meigs syndrome may mimic advanced ovarian cancer. Ultrasound Obstet Gynecol 32:237-238, 2008. [PubMed: 18618418]
- 155.
- Wong LY, Diamond TH: Severe ophthalmopathy developing after treatment of coexisting malignant struma ovarii and Graves' disease. Thyroid 19:1125-1127, 2009. [PubMed: 19772400]
- 156.
- Joja I, Asakawa T, Mitsumori A, et al.: I-123 uptake in nonfunctional struma ovarii. Clin Nucl Med 23:10-12, 1998. [PubMed: 9442957]
- 157.
- Wolff EF, Hughes M, Merino MJ, et al.: Expression of benign and malignant thyroid tissue in ovarian teratomas and the importance of multimodal management as illustrated by a BRAF-positive follicular variant of papillary thyroid cancer. Thyroid 20:981-987, 2010. [PMC free article: PMC2964358] [PubMed: 20718682]
- 158.
- McGill JF, Sturgeon C, Angelos P: Metastatic struma ovarii treated with total thyroidectomy and radioiodine ablation. Endocr Pract 15:167-173, 2009. [PubMed: 19289330]
- 159.
- Ezon I, Zilbert N, Pinkney L, et al.: A large struma ovarii tumor removed via laparoscopy in a 16-year-old adolescent. J Pediatr Surg 42:E19-22, 2007. [PubMed: 17706482]
- 160.
- Roti E, Uberti ED: Iodine excess and hyperthyroidism. Thyroid 11:493-500, 2001. [PubMed: 11396708]
- 161.
- B�rgi H: Iodine excess. Best Pract Res Clin Endocrinol Metab 24:107-115, 2010. [PubMed: 20172475]
- 162.
- Coindet J: Nouvelles recherches sur les effets de l'iode, et sur les pr�cautions � suivre dans le tra�tment du goitre par ce nouveau rem�de. Ann Chimie Phys (Paris) 16:252-266, 1821.
- 163.
- Hennemann G: Historical aspects about the development of our knowledge of morbus Basedow. J Endocrinol Invest 14:617-624, 1991. [PubMed: 1940068]
- 164.
- Fradkin JE, Wolff J: Iodide-induced thyrotoxicosis. Medicine (Baltimore) 62:1-20, 1983. [PubMed: 6218369]
- 165.
- Herrmann J, Kruskemper HL: Gef�hrdung von Patienten mit latenter und manifester Hyperthyreose durch jodhaltige R�ntgenkontrastmittel und Medikamente. Dtsch Med Wochenschr 103:1434, 1437-1443, 1978. [PubMed: 688870]
- 166.
- Hintze G, Blombach O, Fink H, et al.: Risk of iodine-induced thyrotoxicosis after coronary angiography: an investigation in 788 unselected subjects. Eur J Endocrinol 140:264-267, 1999. [PubMed: 10216523]
- 167.
- Martin FI, Tress BW, Colman PG, et al.: Iodine-induced hyperthyroidism due to nonionic contrast radiography in the elderly. Am J Med 95:78-82, 1993. [PubMed: 8328500]
- 168.
- Dietary Reference Intakes: recommended intakes for individuals. . In. 2010 ed: National Academy of Sciences. Institute of Medicine. Food and Nutrition Board2010.
- 169.
- Patrick L: Iodine: deficiency and therapeutic considerations. Altern Med Rev 13:116-127, 2008. [PubMed: 18590348]
- 170.
- Kopp P: Thyroid hormone synthesis: thyroid iodine metabolism. In: Braverman L, Utiger R eds. Werner and Ingbar's the thyroid: a fundamental and clinical text. 9 ed. Philadelphia: Lippincott, Williams & Wilkins; 52-76, 2005.
- 171.
- Wolff J, Chaikoff I, Goldberg R, et al.: The temporary nature of the inhibitory action of excess iodide on organic iodide synthesis in the normal thyroid. Endocrinology 45:504-513, 1949. [PubMed: 15396709]
- 172.
- B�rgi H, Supersaxo Z, Selz B: Iodine deficiency diseases in Switzerland one hundred years after Theodor Kocher's survey: a historical review with some new goitre prevalence data. Acta Endocrinol (Copenh) 123:577-590, 1990. [PubMed: 2284884]
- 173.
- Connolly RJ, Vidor GI, Stewart JC: Increase in thyrotoxicosis in endemic goitre area after iodation of bread. Lancet 1:500-502, 1970. [PubMed: 4190182]
- 174.
- Stewart JC, Vidor GI: Thyrotoxicosis induced by iodine contamination of food--a common unrecognised condition? Br Med J 1:372-375, 1976. [PMC free article: PMC1638791] [PubMed: 946162]
- 175.
- Stanbury JB, Ermans AE, Bourdoux P, et al.: Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid 8:83-100, 1998. [PubMed: 9492158]
- 176.
- Todd CH, Allain T, Gomo ZA, et al.: Increase in thyrotoxicosis associated with iodine supplements in Zimbabwe. Lancet 346:1563-1564, 1995. [PubMed: 7491075]
- 177.
- Bourdoux PP, Ermans AM, Mukalay wa Mukalay A, et al.: Iodine-induced thyrotoxicosis in Kivu, Zaire. Lancet 347:552-553, 1996. [PubMed: 8596306]
- 178.
- Teng W, Shan Z, Teng X, et al.: Effect of iodine intake on thyroid diseases in China. N Engl J Med 354:2783-2793, 2006. [PubMed: 16807415]
- 179.
- Yang F, Shan Z, Teng X, et al.: Chronic iodine excess does not increase the incidence of hyperthyroidism: a prospective community-based epidemiological survey in China. Eur J Endocrinol 156:403-408, 2007. [PubMed: 17389453]
- 180.
- Baltisberger BL, Minder CE, Burgi H: Decrease of incidence of toxic nodular goitre in a region of Switzerland after full correction of mild iodine deficiency. Eur J Endocrinol 132:546-549, 1995. [PubMed: 7749493]
- 181.
- Braverman LE, Woeber KA, Ingbar SH: Induction of myxedema by iodide in patients euthyroid after radioiodin or surgical treatment of diffuse toxic goiter. N Engl J Med 281:816-821, 1969. [PubMed: 4980221]
- 182.
- Clark OH, Cavalieri RR, Moser C, et al.: Iodide-induced hypothyroidism in patients after thyroid resection. Eur J Clin Invest 20:573-580, 1990. [PubMed: 2127746]
- 183.
- Braverman LE, Ingbar SH, Vagenakis AG, et al.: Enhanced susceptibility to iodide myxedema in patients with Hashimoto's disease. J Clin Endocrinol Metab 32:515-521, 1971. [PubMed: 4926257]
- 184.
- Roti E, Minelli R, Gardini E, et al.: Impaired intrathyroidal iodine organification and iodine-induced hypothyroidism in euthyroid women with a previous episode of postpartum thyroiditis. J Clin Endocrinol Metab 73:958-963, 1991. [PubMed: 1658032]
- 185.
- Chow CC, Phillips DI, Lazarus JH, et al.: Effect of low dose iodide supplementation on thyroid function in potentially susceptible subjects: are dietary iodide levels in Britain acceptable? Clin Endocrinol (Oxf) 34:413-416, 1991. [PubMed: 2060151]
- 186.
- Leger AF, Massin JP, Laurent MF, et al.: Iodine-induced thyrotoxicosis: analysis of eighty-five consecutive cases. Eur J Clin Invest 14:449-455, 1984. [PubMed: 6441722]
- 187.
- Ermans AM, Camus M: Modifications of thyroid function induced by chronic administration of iodide in the presence of "autonomous" thyroid tissue. Acta Endocrinol (Copenh) 70:463-475, 1972. [PubMed: 4114611]
- 188.
- Corvilain B, Van Sande J, Dumont JE, et al.: Autonomy in endemic goiter. Thyroid 8:107-113, 1998. [PubMed: 9492160]
- 189.
- Vagenakis AG, Wang CA, Burger A, et al.: Iodide-induced thyrotoxicosis in Boston. N Engl J Med 287:523-527, 1972. [PubMed: 5050425]
- 190.
- B�rgi H, Kohler M, Morselli B: Thyrotoxicosis incidence in Switzerland and benefit of improved iodine supply. Lancet 352:1034, 1998. [PubMed: 9759751]
- 191.
- Lewinski A, Zygmunt A, Karbownik-Lewinska M, et al.: Detrimental effects of increasing iodine supply: iodine-induced hyperthyroidism, following iodine prophylaxis. In: Preedy V, Burrow G, Watson R eds. Comprehensive handbook of iodine. Amsterdam: Elsevier; 871�875, 2009.
- 192.
- Teng X, Shan Z, Teng W, et al.: Experimental study on the effects of chronic iodine excess on thyroid function, structure, and autoimmunity in autoimmune-prone NOD.H-2h4 mice. Clin Exp Med 9:51-59, 2009. [PubMed: 18953634]
- 193.
- Many MC, Mestdagh C, van den Hove MF, et al.: In vitro study of acute toxic effects of high iodide doses in human thyroid follicles. Endocrinology 131:621-630, 1992. [PubMed: 1639011]
- 194.
- B�low Pedersen I, Laurberg P, Knudsen N, et al.: Increase in incidence of hyperthyroidism predominantly occurs in young people after iodine fortification of salt in Denmark. J Clin Endocrinol Metab 91:3830-3834, 2006. [PubMed: 16849408]
- 195.
- Bastemir M, Emral R, Erdogan G, et al.: High prevalence of thyroid dysfunction and autoimmune thyroiditis in adolescents after elimination of iodine deficiency in the Eastern Black Sea Region of Turkey. Thyroid 16:1265-1271, 2006. [PubMed: 17199437]
- 196.
- Camargo RY, Tomimori EK, Neves SC, et al.: Thyroid and the environment: exposure to excessive nutritional iodine increases the prevalence of thyroid disorders in Sao Paulo, Brazil. Eur J Endocrinol 159:293-299, 2008. [PubMed: 18586897]
- 197.
- Zimmermann MB, Moretti D, Chaouki N, et al.: Introduction of iodized salt to severely iodine-deficient children does not provoke thyroid autoimmunity: a one-year prospective trial in northern Morocco. Thyroid 13:199-203, 2003. [PubMed: 12699595]
- 198.
- Savoie JC, Massin JP, Thomopoulos P, et al.: Iodine-induced thyrotoxicosis in apparently normal thyroid glands. J Clin Endocrinol Metab 41:685-691, 1975. [PubMed: 1176580]
- 199.
- Bogazzi F, Bartalena L, Gasperi M, et al.: The various effects of amiodarone on thyroid function. Thyroid 11:511-519, 2001. [PubMed: 11396710]
- 200.
- Bogazzi F, Bartalena L, Martino E: Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab 95:2529-2535, 2010. [PubMed: 20525904]
- 201.
- Basaria S, Cooper DS: Amiodarone and the thyroid. Am J Med 118:706-714, 2005. [PubMed: 15989900]
- 202.
- Martino E, Aghini-Lombardi F, Bartalena L, et al.: Enhanced susceptibility to amiodarone-induced hypothyroidism in patients with thyroid autoimmune disease. Arch Intern Med 154:2722-2726, 1994. [PubMed: 7993156]
- 203.
- Martino E, Bartalena L, Bogazzi F, et al.: The effects of amiodarone on the thyroid. Endocr Rev 22:240-254, 2001. [PubMed: 11294826]
- 204.
- Bogazzi F, Bartalena L, Brogioni S, et al.: Color flow Doppler sonography rapidly differentiates type I and type II amiodarone-induced thyrotoxicosis. Thyroid 7:541-545, 1997. [PubMed: 9292940]
- 205.
- Bogazzi F, Martino E, Dell'Unto E, et al.: Thyroid color flow doppler sonography and radioiodine uptake in 55 consecutive patients with amiodarone-induced thyrotoxicosis. J Endocrinol Invest 26:635-640, 2003. [PubMed: 14594114]
- 206.
- Cappiello E, Boldorini R, Tosoni A, et al.: Ultrastructural evidence of thyroid damage in amiodarone-induced thyrotoxicosis. J Endocrinol Invest 18:862-868, 1995. [PubMed: 8778159]
- 207.
- Ross DS: Syndromes of thyrotoxicosis with low radioactive iodine uptake. Endocrinol Metab Clin North Am 27:169-185, 1998. [PubMed: 9534035]
- 208.
- Lazarus J: Sporadic and postpartum thyroiditis. In: Braverman L, Utiger R eds. The Thyroid. Philadelphia: Lippincott-Raven; 524-535, 2005.
- 209.
- Dorfman SG, Cooperman MT, Nelson RL, et al.: Painless thyroiditis and transient hyperthyroidism without goiter. Ann Intern Med 86:24-28, 1977. [PubMed: 576376]
- 210.
- Jackson IM: Editorial: "Hyper-thyroiditis" - a diagnostic pitfall. N Engl J Med 293:661-662, 1975. [PubMed: 1173936]
- 211.
- Nikolai TF, Brosseau J, Kettrick MA, et al.: Lymphocytic thyroiditis with spontaneously resolving hyperthyroidism (silent thyroiditis). Arch Intern Med 140:478-482, 1980. [PubMed: 6892676]
- 212.
- Woolf PD, Daly R: Thyrotoxicosis with painless thyroiditis. Am J Med 60:73-79, 1976. [PubMed: 56130]
- 213.
- Woolf PD: Transient painless thyroiditis with hyperthyroidism: a variant of lymphocytic thyroiditis? Endocr Rev 1:411-420, 1980. [PubMed: 7018893]
- 214.
- Gegick CG, Herring WB: Painless subacute thyroiditis: a report of two cases. N C Med J 38:387-389, 1977. [PubMed: 267808]
- 215.
- Hamburger JI: Occult subacute thyroiditis--diagnostic challenge. Mich Med 70:1125-1127, 1971. [PubMed: 5118969]
- 216.
- Morrison J, Caplan RH: Typical and atypical ('silent') subacute thyroiditis in a wife and husband. Arch Intern Med 138:45-48, 1978. [PubMed: 619831]
- 217.
- Gorman CA, Duick DS, Woolner LB, et al.: Transient hyperthyroidism in patients with lymphocytic thyroiditis. Mayo Clin Proc 53:359-365, 1978. [PubMed: 580628]
- 218.
- Vitug AC, Goldman JM: Silent (painless) thyroiditis. Evidence of a geographic variation in frequency. Arch Intern Med 145:473-475, 1985. [PubMed: 3838433]
- 219.
- Schneeberg NG: Silent thyroiditis. Arch Intern Med 143:2214, 1983. [PubMed: 6639248]
- 220.
- Hedberg CW, Fishbein DB, Janssen RS, et al.: An outbreak of thyrotoxicosis caused by the consumption of bovine thyroid gland in ground beef. N Engl J Med 316:993-998, 1987. [PubMed: 3561455]
- 221.
- Kinney JS, Hurwitz ES, Fishbein DB, et al.: Community outbreak of thyrotoxicosis: epidemiology, immunogenetic characteristics, and long-term outcome. Am J Med 84:10-18, 1988. [PubMed: 3257352]
- 222.
- Iitaka M, Morgenthaler NG, Momotani N, et al.: Stimulation of thyroid-stimulating hormone (TSH) receptor antibody production following painless thyroiditis. Clin Endocrinol (Oxf) 60:49-53, 2004. [PubMed: 14678287]
- 223.
- Miyakawa M, Tsushima T, Onoda N, et al.: Thyroid ultrasonography related to clinical and laboratory findings in patients with silent thyroiditis. J Endocrinol Invest 15:289-295, 1992. [PubMed: 1512420]
- 224.
- Nikolai TF, Coombs GJ, McKenzie AK, et al.: Treatment of lymphocytic thyroiditis with spontaneously resolving hyperthyroidism (silent thyroiditis). Arch Intern Med 142:2281-2283, 1982. [PubMed: 6897348]
- 225.
- Nikolai TF, Coombs GJ, McKenzie AK: Lymphocytic thyroiditis with spontaneously resolving hyperthyroidism and subacute thyroiditis. Long-term follow-up. Arch Intern Med 141:1455-1458, 1981. [PubMed: 7283556]
- 226.
- Tokuda Y, Kasagi K, Iida Y, et al.: Sonography of subacute thyroiditis: changes in the findings during the course of the disease. J Clin Ultrasound 18:21-26, 1990. [PubMed: 2152779]
- 227.
- Parker M, Klein I, Fishman LM, et al.: Silent thyrotoxic thyroiditis in association with chronic adrenocortical insufficiency. Arch Intern Med 140:1108-1109, 1980. [PubMed: 7396620]
- 228.
- Muller AF, Drexhage HA, Berghout A: Postpartum thyroiditis and autoimmune thyroiditis in women of childbearing age: recent insights and consequences for antenatal and postnatal care. Endocr Rev 22:605-630, 2001. [PubMed: 11588143]
- 229.
- Mizukami Y, Michigishi T, Hashimoto T, et al.: Silent thyroiditis: a histologic and immunohistochemical study. Hum Pathol 19:423-431, 1988. [PubMed: 3284807]
- 230.
- Roberts CG, Ladenson PW: Hypothyroidism. Lancet 363:793-803, 2004. [PubMed: 15016491]
- 231.
- Cohen JH, 3rd, Ingbar SH, Braverman LE: Thyrotoxicosis due to ingestion of excess thyroid hormone. Endocr Rev 10:113-124, 1989. [PubMed: 2666114]
- 232.
- Biondi B, Cooper DS: Benefits of thyrotropin suppression versus the risks of adverse effects in differentiated thyroid cancer. Thyroid 20:135-146, 2010. [PubMed: 20151821]
- 233.
- Biondi B, Fazio S, Carella C, et al.: Cardiac effects of long term thyrotropin-suppressive therapy with levothyroxine. J Clin Endocrinol Metab 77:334-338, 1993. [PubMed: 8345037]
- 234.
- Shapiro LE, Sievert R, Ong L, et al.: Minimal cardiac effects in asymptomatic athyreotic patients chronically treated with thyrotropin-suppressive doses of L-thyroxine. J Clin Endocrinol Metab 82:2592-2595, 1997. [PubMed: 9253339]
- 235.
- Faber J, Galloe AM: Changes in bone mass during prolonged subclinical hyperthyroidism due to L-thyroxine treatment: a meta-analysis. Eur J Endocrinol 130:350-356, 1994. [PubMed: 8162163]
- 236.
- Franklyn J, Betteridge J, Holder R, et al.: Bone mineral density in thyroxine treated females with or without a previous history of thyrotoxicosis. Clin Endocrinol (Oxf) 41:425-432, 1994. [PubMed: 7955453]
- 237.
- Bogazzi F, Bartalena L, Scarcello G, et al.: The age of patients with thyrotoxicosis factitia in Italy from 1973 to 1996. J Endocrinol Invest 22:128-133, 1999. [PubMed: 10195380]
- 238.
- Feit S, Feit H: Thyrotoxicosis factitia veterinarius. Ann Intern Med 127:168, 1997. [PubMed: 9230020]
- 239.
- Cavalieri RR, Gerard SK: Unusual types of thyrotoxicosis. Adv Intern Med 36:271-286, 1991. [PubMed: 1850947]
- 240.
- Mandel SH, Magnusson AR, Burton BT, et al.: Massive levothyroxine ingestion. Conservative management. Clin Pediatr (Phila) 28:374-376, 1989. [PubMed: 2758719]
- 241.
- Mariotti S, Martino E, Cupini C, et al.: Low serum thyroglobulin as a clue to the diagnosis of thyrotoxicosis factitia. N Engl J Med 307:410-412, 1982. [PubMed: 7088115]
- 242.
- Bouillon R, Verresen L, Staels F, et al.: The measurement of fecal thyroxine in the diagnosis of thyrotoxicosis factitia. Thyroid 3:101-103, 1993. [PubMed: 8369649]
- 243.
- Bogazzi F, Bartalena L, Brogioni S, et al.: Thyroid vascularity and blood flow are not dependent on serum thyroid hormone levels: studies in vivo by color flow doppler sonography. Eur J Endocrinol 140:452-456, 1999. [PubMed: 10229913]
- INTRODUCTION
- TOXIC ADENOMA
- TOXIC MULTINODULAR GOITER
- HYPERTHYROID THYROID CARCINOMA
- FAMILIAL NON-AUTOIMMUNE HYPERTHYROIDISM WITH TSHR MUTATIONS
- SPORADIC NON-AUTOIMMUNE HYPERTHYROIDISM
- TSH-SECRETING PITUITARY ADENOMA
- RESISTANCE TO THYROID HORMONE
- HCG-INDUCED GESTATIONAL HYPERTHYROIDISM
- FAMILIAL HYPERSENSITIVITY TO HCG
- TROPHOBLAST TUMORS: HYDATIFORM MOLES AND CHORIOCARCINOMA
- STRUMA OVARII
- IODINE-INDUCED HYPERTHYROIDISM
- I. IODINE-INDUCED THYROTOXICOSIS
- II. AMIODARONE INDUCED THYROID DISEASE
- THYROIDITIS
- I. ACUTE OR SUBACUTE (DE QUERVAIN'S) THYROIDITIS
- II. SILENT OR PAINLESS THYROIDITIS
- III. POSTPARTUM THYROIDITIS
- IV. HASHIMOTO'S THYROIDITIS
- THYROTOXICOSIS FACTITIA
- SUMMARY
- ACKNOWLEDGMENT
- Review Hyperthyroidism.[J Indian Med Assoc. 2006]Review Hyperthyroidism.Maji D. J Indian Med Assoc. 2006 Oct; 104(10):563-4, 566-7.
- Thyrotoxic adenoma followed by atypical hyperthyroidism due to struma ovarii: clinical and genetic studies.[Eur J Endocrinol. 2004]Thyrotoxic adenoma followed by atypical hyperthyroidism due to struma ovarii: clinical and genetic studies.Ciccarelli A, Valdes-Socin H, Parma J, Khoo SK, Schoumans J, Colao A, Hamoir E, Beckers A. Eur J Endocrinol. 2004 Apr; 150(4):431-7.
- Review The Role of Nuclear Medicine in the Clinical Management of Benign Thyroid Disorders, Part 1: Hyperthyroidism.[J Nucl Med. 2021]Review The Role of Nuclear Medicine in the Clinical Management of Benign Thyroid Disorders, Part 1: Hyperthyroidism.Mariani G, Tonacchera M, Grosso M, Orsolini F, Vitti P, Strauss HW. J Nucl Med. 2021 Mar; 62(3):304-312. Epub 2020 Oct 2.
- Review Iodine excess and hyperthyroidism.[Thyroid. 2001]Review Iodine excess and hyperthyroidism.Roti E, Uberti ED. Thyroid. 2001 May; 11(5):493-500.
- [Agranulocytosis and acute coronary syndrome in apathetic hyperthyroidism].[Srp Arh Celok Lek. 2003][Agranulocytosis and acute coronary syndrome in apathetic hyperthyroidism].Ivović M, Radiojković B, Penezić Z, Stojković M, Tancić M, Vujović S, Bogdanović A, Drezgić M. Srp Arh Celok Lek. 2003 May-Jun; 131(5-6):249-53.
- Thyrotoxicosis of other Etiologies - EndotextThyrotoxicosis of other Etiologies - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...