

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Vitamin D production in the skin under the influence of sunlight (UVB) is maximized at levels of sunlight exposure that do not burn the skin. Further metabolism of vitamin D to its major circulating form (25(OH)D) and hormonal form (1,25(OH)2D) takes place in the liver and kidney, respectively, but also in other tissues where the 1,25(OH)2D produced serves a paracrine/autocrine function: examples include the skin, cells of the immune system, parathyroid gland, intestinal epithelium, prostate, and breast. Parathyroid hormone, FGF23, calcium and phosphate are the major regulators of the renal 1-hydroxylase (CYP27B1, the enzyme producing 1,25(OH)2D); regulation of the extra renal 1-hydroxylase differs from that in the kidney and involves cytokines. The major enzyme that catabolizes 25(OH)D and 1,25(OH)2D is the 24-hydroxylase; like the 1-hydroxylase it is tightly controlled in the kidney in a manner opposite to that of the 1-hydroxylase, but like the 1-hydroxylase it is widespread in other tissues where its regulation is different from that of the kidney. Vitamin D and its metabolites are carried in the blood bound to vitamin D binding protein (DBP) and albumin--for most tissues it is the free (i.e., unbound) metabolite that enters the cell; however, DBP bound metabolites can enter some cells such as the kidney and parathyroid gland through a megalin/cubilin mechanism. Most but not all actions of 1,25(OH)2D are mediated by the vitamin D receptor (VDR). VDR is a transcription factor that partners with other transcription factors such as retinoid X receptor that when bound to 1,25(OH)2D regulates gene transcription either positively or negatively depending on other cofactors to which it binds or interacts. The VDR is found in most cells, not just those involved with bone and mineral homeostasis (i.e., bone, gut, kidney) resulting in wide spread actions of 1,25(OH)2D on most physiologic and pathologic processes. Animal studies indicate that vitamin D has beneficial effects on various cancers, blood pressure, heart disease, immunologic disorders, but these non-skeletal effects have been difficult to prove in humans in randomized controlled trials. Analogs of 1,25(OH)2D are being developed to achieve specificity for non-skeletal target tissues such as the parathyroid gland and cancers to avoid the hypercalcemia resulting from 1,25(OH)2D itself. The level of vitamin D intake and achieved serum levels of 25(OH)D that are optimal and safe for skeletal health and the non-skeletal actions remain controversial, but are likely between an intake of 800-2000IU vitamin D in the diet and 20-50ng/ml 25(OH)D in the blood. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
OVERVIEW
Rickets became a public health problem with the movement of the population from the farms to the cities during the Industrial Revolution. Various foods such as cod liver oil and irradiation of other foods including plants were found to prevent or cure this disease, leading eventually to the discovery of the active principle—vitamin D. Vitamin D comes in two forms (D2 and D3) which differ chemically in their side chains. These structural differences alter their binding to the carrier protein vitamin D binding protein (DBP) and their metabolism, but in general the biologic activity of their active metabolites is comparable. Vitamin D3 is produced in the skin from 7-dehydrocholesterol by UV irradiation, which breaks the B ring to form pre-D3. Pre-D3 isomerizes to D3 but with continued UV irradiation to tachysterol and lumisterol. D3 is preferentially removed from the skin, bound to DBP. The liver and other tissues metabolize vitamin D, whether from the skin or oral ingestion, to 25OHD, the principal circulating form of vitamin D. Several enzymes have 25-hydroxylase activity, but CYP2R1 is the most important. 25OHD is then further metabolized to 1,25(OH)2D principally in the kidney, by the enzyme CYP27B1, although other tissues including various epithelial cells, cells of the immune system, and the parathyroid gland contain this enzyme. 1,25(OH)2D is the principal hormonal form of vitamin D, responsible for most of its biologic actions. The production of 1,25(OH)2D in the kidney is tightly controlled, being stimulated by parathyroid hormone (PTH), and inhibited by calcium, phosphate and FGF23. Extrarenal production of 1,25(OH)2D as in keratinocytes and macrophages is under different control, being stimulated primarily by cytokines such as tumor necrosis factor alfa (TNFα) and interferon gamma (IFNg). 1,25(OH)2D reduces 1,25(OH)2D levels in cells primarily by stimulating its catabolism through the induction of CYP24A1, the 24-hydroxylase. 25OHD and 1,25(OH)2D are hydroxylated in the 24 position by this enzyme to form 24,25(OH)2D and 1,24,25(OH)3D, respectively. This 24-hydroxylation is generally the first step in the catabolism of these active metabolites to the final end product of calcitroic acid, although 24,25(OH)2D and 1,24,25(OH)3D have their own biologic activities. CYP24A1 also has 23-hydroxylase activity that leads to a different end product. Different species differ in their ratio of 23-hydroxylase/24-hydroxyase activity in their CYP24A1 enzyme, but in humans the 24-hydroxyase activity predominates. Like CYP27B1, CYP24A1 is widely expressed. CYP24A1 is induced by 1,25(OH)2D in most tissues, which serves as an important feedback mechanism to avoid vitamin D toxicity. In the kidney, PTH inhibits CYP24A1, whereas FGF23, calcium and phosphate stimulates it, just the opposite of the actions of these hormones and minerals on CYP27B1. However, such regulation is not seen in other tissues. In macrophages, CYP24A1 is either missing or defective, so in situations such as granulomatous diseases like sarcoidosis in which macrophage production of 1,25(OH)2D is increased, hypercalcemia and hypercalciuria due to elevated 1,25(OH)2D can occur without the counter regulation by CYP24A1.
The vitamin D metabolites are transported in blood bound to DBP and albumin. Very little circulates as the free form. The liver produces DBP and albumin, production that is decreased in liver disease, and these proteins may be lost in protein losing enteropathies or the nephrotic syndrome. Thus, individuals with liver, intestinal or renal diseases which result in low levels of these transport proteins may have low total levels of the vitamin D metabolites without necessarily being vitamin D deficient as their free concentrations may be normal.
The receptor for 1,25(OH)2D (VDR) is a transcription factor regulating the expression of genes which mediate its biologic activity. VDR is a member of a rather large family of nuclear hormone receptors which includes the receptors for glucocorticoids, mineralocorticoids, sex hormones, thyroid hormone, and vitamin A metabolites or retinoids. The VDR is widely distributed, and is not restricted to those tissues considered the classic target tissues of vitamin D. The VDR upon binding to 1,25(OH)2D heterodimerizes with other nuclear hormone receptors, in particular the family of retinoid X receptors. This complex then binds to special DNA sequences called vitamin D response elements (VDRE) generally within the genes it regulates, although these VDREs can be thousands of base pairs from the transcription start site. There are thousands of the VDREs in hundreds of genes, and the profile of active VDREs (and regulated genes) varies from cell to cell. A variety of additional proteins called coregulators complex with the VDR to activate (coactivators) or inhibit (corepressors) VDR transcriptional activity. Coactivator factors involved in VDR mediated transcription include factors with histone acetylase activity, including steroid receptor coactivator (SRC) 1, SRC 2 and SRC 3, and CREB-binding protein p300, in addition to the SWI–SNF ATP dependent chromatin remodeling complex, methyltransferases and the Mediator complex (aka DRIP), which functions to recruit RNA polymerases. VDR binding sites are associated with sites for other transcription factors such as p63, C–EBPα, C–EBPβ, Runx2 and PU.1, which can cooperate with VDR and VDR coregulators to influence 1,25(OH)2D responses in target cells. Among other functions these coregulators reconfigure the chromatin structure to bring the VDR/VDRE to the transcription start site, explaining how such distant VDR/VDREs can regulate gene transcription. In addition to coactivators there are a number of corepressors. One such corepressor of VDR action in the skin is called hairless, in that its loss or mutation, like that of the VDR, leads to altered hair follicle cycling resulting in baldness. Corepressors typically work by recruiting histone deacetylases (HDAC) or methyl transferases (MT) to the gene which reverses the actions of HAT, leading to a reduction in access to the gene by the transcription machinery. These coregulators can be specific for different genes, and different cells differentially express these coregulators, providing some specificity for the actions of 1,25(OH)2D and VDR.
In addition to regulating gene expression, 1,25(OH)2D has a number of non-genomic actions including the ability to stimulate calcium transport across the plasma membrane. The mechanisms mediating these non-genomic actions and their physiologic significance remain unclear. Similarly, it is not clear that all actions of the VDR require the ligand 1,25(OH)2D. The best example of this is the hair loss in animals and subjects with VDR mutations but not in animals and subjects with mutations in CYP27B1, the enzyme producing 1,25(OH)2D. As mentioned, the VDR is widely distributed, and the actions of 1,25(OH)2D are quite varied. The classic target tissues—bone, gut, and kidney—are involved with calcium homeostasis. The mechanisms by which 1,25(OH)2D regulates transcellular calcium transport are best understood in the intestine. Here 1,25(OH)2D stimulates calcium entry across the brush border membrane into the cell, transport of calcium through the cell, and removal of calcium from the cell at the basolateral membrane. Calcium entry at the brush border membrane occurs down a steep electrochemical gradient. It is controlled in large measure by a specific calcium channel called TRPV6 and in humans also by a homologous calcium channel TRPV5. Transport of calcium through the cell is regulated by a class of calcium binding proteins called calbindins. Much of the transport occurs within vesicles that form in the terminal web. Removal of calcium from the cell at the basolateral membrane requires energy and is mediated by the ATP requiring calcium pump or CaATPase (PMCA1b) as well as the sodium/calcium exchange protein (NCX1). 1,25(OH)2D induces TRPV6 and TRPV5, the calbindins, and the CaATPase, but not all aspects of transcellular calcium transport are a function of new protein synthesis. Animals null for calbindin 9k (the major calbindin in mammalian intestine) have little impairment of intestinal calcium transport. Animals null for TRPV6, on the other hand, have a reduction in intestinal calcium transport, but the deficit is not profound. Thus, it is likely that compensatory mechanisms for intestinal calcium transport exist that have yet to be discovered. Similar mechanisms mediate 1,25(OH)2D regulated calcium reabsorption in the distal tubule of the kidney. The proteins involved are homologous but not identical (TRPV5 and Calbindin 28k, for example). The situation in bone, however, is less clear. VDR are found in osteoblasts, the bone forming cells. 1,25(OH)2D promotes the differentiation of osteoblasts and regulates the production of proteins such as collagen, alkaline phosphatase, and osteocalcin thought to be important in bone formation. 1,25(OH)2D also induces RANKL, a membrane bound protein in osteoblasts that enables osteoblasts to stimulate the formation and activity of osteoclasts. Thus 1,25(OH)2D regulates both bone formation and bone resorption. Some evidence suggests that the major effect of 1,25(OH)2D on bone is to provide adequate levels of calcium and phosphate from the intestine. The rickets of patients with a mutated VDR or of mice in which the VDR has been deleted can be prevented/corrected by normalizing serum calcium and phosphate levels by dietary means. On the other hand, normal bone formation is not restored, and with time the VDR null mice become osteoporotic despite the high calcium/phosphate diet. Moreover, the VDR in osteoblasts/osteocytes appears to control bone resorption especially when dietary calcium is limited. Whether subjects with VDR mutations also develop osteoporosis prematurely or fail to maintain serum calcium in times of calcium deficiency has not been reported.
The non-classic actions of 1,25(OH)2D include regulation of cellular proliferation and differentiation, regulation of hormone secretion, and regulation of immune function. The ability of 1,25(OH)2D to inhibit proliferation and stimulate differentiation has led to the development of a number of analogs in the hopes of treating hyperproliferative disorders such as psoriasis and cancer without raising serum calcium. Psoriasis is now successfully treated with several vitamin D analogs. Observational studies are promising with respect to adequate vitamin D nutrition and cancer prevention. However, supplementation with vitamin D of subjects with adequate vitamin D levels to start with has not been shown to decrease cancer incidence but may be beneficial for cancer mortality. 1,25(OH)2D inhibits parathyroid hormone secretion and stimulates insulin secretion. A number of analogs and 1,25(OH)2D itself are currently available for use in the treatment of secondary hyperparathyroidism accompanying renal failure. Epidemiologic evidence indicates that vitamin D deficiency is associated with increased risk of both type 1 and type 2 diabetes mellitus, but prospective clinical trials to demonstrate a role for vitamin D supplementation in preventing the conversion of prediabetes to diabetes has not shown benefit in vitamin D replete individuals. However, there may be benefit in vitamin D deficient patients. The ability of 1,25(OH)2D to regulate immune function is likely part of its efficacy in the treatment of psoriasis. A number of other autoimmune diseases have been found in animal studies to respond favorably to vitamin D and 1,25(OH)2D or its analogs, and epidemiologic evidence linking vitamin D deficiency to increased incidence of these diseases has been reported. Similarly, epidemiologic evidence linking vitamin D deficiency to a number of respiratory illnesses is substantial, including increased risk of COVID-19 infections.
DISCOVERY
The first clear description of rickets was by Whistler (1) in 1645. However, it was not until the Industrial Revolution with the mass movement of the population from the farms to the smoke- filled cities that rickets became a public health problem, most notably in England where sunlight intensity was already marginal for much of the year. Mellanby (2) in Great Britain and McCollum (3) in the United States developed animal models for rickets and showed that rickets could be cured with cod liver oil. McCollum heated the cod liver oil to destroy its vitamin A content and found that it still had antirachitic properties; he named the antirachitic factor vitamin D. Steenbock and Black (4) then demonstrated that UV irradiation of food, in particular non saponifiable lipids, could treat rickets. Meanwhile, clinical investigations revealed that rickets could be prevented or cured in children with sunlight or artificial UV exposure (5,6) suggesting that what subsequently became known as vitamin D could be produced by irradiation of precursors in vivo. Ultimately, Askew et al. (7) isolated and determined the structure of vitamin D2 (ergocalciferol) from irradiated plant sterols (ergosterol), and Windaus et al. (8) determined the structures and pathway by which 7-dehydrocholesterol (7-DHC) in the skin is converted to vitamin D3 (cholecalciferol). The name vitamin D1 refers to what proved to be an error of an earlier identification, and is not used. The structures and pathways of production of vitamin D3 are shown in figure 1. The structures of vitamins D2 and D3 differ in the side chain where D2 contains a double bond (C22-23) and an additional methyl group attached to C24. In this chapter the designation of D will refer to both D3 and D2.
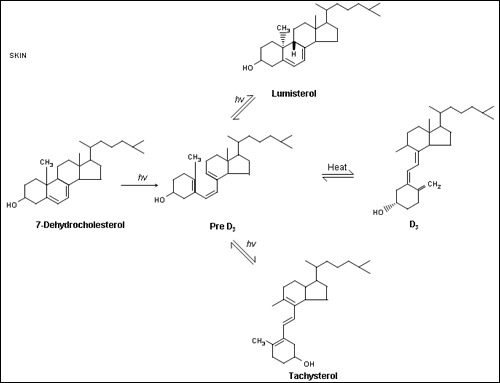
Figure 1.
The production of vitamin D3 from 7-dehydrocholesterol in the epidermis. Sunlight (the ultraviolet B component) breaks the B ring of the cholesterol structure to form pre- D3. Pre-D3 then undergoes a thermal induced rearrangement to form D3. Continued irradiation of pre- D3 leads to the reversible formation of lumisterol3 and tachysterol3 which can revert back to pre-D3 in the dark.
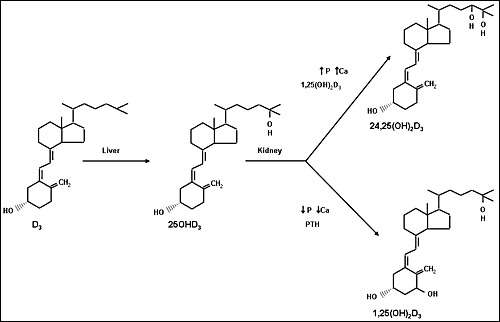
Figure 2.
The metabolism of vitamin D. The liver converts vitamin D to 25OHD. The kidney converts 25OHD to 1,25(OH)2D and 24,25(OH)2D. Other tissues contain these enzymes, but the liver is the main source for 25-hydroxylation, and the kidney is the main source for 1α-hydroxylation. Control of metabolism of vitamin D to its active metabolite, 1,25(OH)2D, is exerted primarily at the renal level where calcium, phosphorus, parathyroid hormone, FGF23, and 1,25(OH)2D regulate the levels of 1,25(OH)2D produced.
METABOLISM
Vitamin D3 produced in the epidermis must be further metabolized to be active. The first step, 25-hydroxylation, takes place primarily in the liver, although other tissues have this enzymatic activity as well. As will be discussed below, there are several 25-hydroxylases. 25OHD is the major circulating form of vitamin D. However, in order for vitamin D metabolites to achieve maximum biologic activity they must be further hydroxylated in the 1α position by the enzyme CYP27B1; 1,25(OH)2D is the most potent metabolite of vitamin D and accounts for most of its biologic actions. The 1α hydroxylation occurs primarily in the kidney, although as for the 25-hydroxylase, other tissues have this enzyme. Vitamin D and its metabolites, 25OHD and 1,25(OH)2D, can also be hydroxylated in the 24 position. This may serve to activate the metabolite or analog as 1,25(OH)2D and 1,24(OH)2D have similar biologic potency, and 1,24,25(OH)3D has activity approximately 1/10 that of 1,25(OH)2D. However, 24-hydroxylation of metabolites with an existing 25OH group leads to further catabolism. The details of these reactions are described below.
Cutaneous Production of Vitamin D3
The precursor of vitamin D, 7-dehydrocholesterol (7-DHC) is on the Kandutsch-Russell cholesterol pathway. The final enzymatic reaction mediated by 7-dehyrocholesterol reductase converting 7-DHC to cholesterol is regulated by a number of factors including vitamin D and cholesterol which enhance its degradation thus enabling increased levels of 7-DHC for conversion to vitamin D (9). Although irradiation of 7-DHC was known to produce pre-D3 (which subsequently undergoes a temperature rearrangement of the triene structure to form D3), lumisterol, and tachysterol (figure 1), the physiologic regulation of this pathway was not well understood until the studies of Holick and his colleagues (10-12). They demonstrated that the formation of pre-D3 under the influence of solar or UV irradiation (maximal effective wavelength between 290-310) is relatively rapid and reaches a maximum within hours. UV irradiation further converts pre-D3 to lumisterol and tachysterol. Both the degree of epidermal pigmentation and the intensity of exposure correlate with the time required to achieve this maximal concentration of pre-D3, but do not alter the maximal level achieved. Although pre-D3 levels reach a maximum level, the biologically inactive lumisterol continues to accumulate with continued UV exposure. Tachysterol is also formed, but like pre-D3, does not accumulate with extended UV exposure. The formation of lumisterol is reversible and can be converted back to pre-D3 as pre-D3 levels fall. At 0oC, no D3 is formed; however, at 37oC pre-D3 is slowly converted to D3. Thus, short exposure to sunlight would be expected to lead to a prolonged production of D3 in the exposed skin because of the slow thermal conversion of pre-D3 to D3 and the conversion of lumisterol to pre-D3. Prolonged exposure to sunlight would not produce toxic amounts of D3 because of the photoconversion of pre-D3 to lumisterol and tachysterol as well as the photoconversion of D3 itself to suprasterols I and II and 5,6 transvitamin D3 (13).
Melanin in the epidermis, by absorbing UV irradiation, can reduce the effectiveness of sunlight in producing D3 in the skin. This may be one important reason for the lower 25OHD levels (a well-documented surrogate measure for vitamin D levels in the body) in Blacks and Hispanics living in temperate latitudes (14). Sunlight exposure increases melanin production, and so provides another mechanism by which excess D3 production can be prevented. The intensity of UV irradiation is also important for effective D3 production. The seasonal variation of 25OHD levels can be quite pronounced with higher levels during the summer months and lower levels during the winter. The extent of this seasonal variation depends on the latitude, and thus the intensity of the sunlight striking the exposed skin. In Edmonton, Canada (52oN) very little D3 is produced in exposed skin from mid-October to mid-April; Boston (42oN) has a somewhat longer period for effective D3 production; whereas in Los Angeles (34oN) and San Juan (18oN) the skin is able to produce D3 all year long (15). These findings apply to sea level. At higher elevations there is less atmospheric absorption of UVB, so that skiers can make vitamin D even in winter on sunny days. Peak D3 production occurs around noon, with a larger portion of the day being capable of producing D3 in the skin during the summer than other times of the year. Clothing (16) and sunscreens (17) effectively prevent D3 production in the covered areas. This is one likely explanation for the observation that the Bedouins in the Middle East, who totally cover their bodies with clothing, are more prone to develop rickets and osteomalacia than the Israeli Jews with comparable sunlight exposure.
Hepatic Production of 25OHD
The next step in the bioactivation of D2 and D3, hydroxylation to 25OHD, takes place primarily in the liver although a number of other tissues express this enzymatic activity. 25OHD is the major circulating form of vitamin D and provides a clinically useful marker for vitamin D status. DeLuca and colleagues were the first to identify 25OHD and demonstrate its production in the liver over 30 years ago, but ambiguity remains as to the actual enzyme(s) responsible for this activity. 25-hydroxylase activity has been found in both the liver mitochondria and endoplasmic reticulum, and the enzymatic activities appear to differ indicating different proteins. At this point most attention has been paid to the mitochondrial CYP27A1 and the microsomal CYP2R1. However, in mouse knockout studies and in humans with mutations in these enzymes, only CYP2R1 loss is associated with decreased 25OHD levels (18,19). However, deletion or mutation of CYP2R1 does not totally eliminate 25OHD production These are mixed function oxidases, but differ in apparent Kms and substrate specificities.
The mitochondrial 25-hydroxylase is now well accepted as CYP27A1, an enzyme first identified as catalyzing a critical step in the bile acid synthesis pathway. This is a high capacity, low affinity enzyme consistent with the observation that 25-hydroxylation is not generally rate limiting in vitamin D metabolism. Although initial studies suggested that the vitamin D3-25-hydroxylase and cholestane triol 27-hydroxyase activities in liver mitochondria were due to distinct enzymes with differential regulation, the cloning of CYP27A1 and the demonstration that it contained both activities has put this issue to rest (20-22). CYP27A1 is widely distributed throughout different tissues with highest levels in liver and muscle, but also in kidney, intestine, lung, skin, and bone (20-23). Mutations in CYP27A1 lead to cerebrotendinous xanthomatosis (24,25), and are associated with abnormal vitamin D and/or calcium metabolism in some but not all of these patients (25-27). However, mice in which CYP27A1 is deleted actually have elevated 25OHD levels along with the disruption in bile acid synthesis (28). CYP27A1 can hydroxylate vitamin D and related compounds at the 24, 25, and 27 positions. However, D2 appears to be preferentially 24-hydroxylated, whereas D3 is preferentially 25-hydroxylated (29). The 1αOH derivatives of D are more rapidly hydroxylated than the parent compounds (30). These differences between D2 and D3 and their 1αOH derivatives may explain the differences in biologic activity between D2 and D3 or between 1αOHD2 and 1αOHD3.
The major microsomal 25-hydroxylase is CYP2R1, although other enzymes have been shown in in vitro studies to have 25-hydroxylase activity. This enzyme like that of CYP27A1 is widely distributed, although it is most abundantly expressed in liver, skin and testes (30). Unlike CYP27A1, CYP2R1 25-hydroxylates D2 and D3 equally (30). Several Nigerian families have been shown to have CYP2R1 mutations in family members with rickets (19,31). These subjects respond to D therapy but suboptimally (19,31). Mice lacking CYP2R1 have reduced 25OHD levels, unlike mice lacking CYP27A1, but even the combined deletion of CYP2R1 and CYP27A1 does not reduce these levels more than about 70% (18). Thus, neither CYP27A1 nor CYP2R1 by themselves account for all 25-hydroxyase activity in the body, suggesting a role of other yet to be described 25-hydroxylases.
Studies of the regulation of 25-hydroxylation have not been completely consistent, most likely because of the initial failure to appreciate that at least two enzymatic activities were involved and because of species differences. In general, 25-hydroxylation in the liver is little affected by vitamin D status. However, CYP27A1 expression in the intestine (32) and kidney (33) is reduced by 1,25(OH)2D. Not surprisingly bile acids decrease CYP27A1 expression (34) as does insulin (35) through an unknown mechanism. Dexamethasone, on the other hand, increases CYP27A1 expression (36). CYP2R1 appears to be mediated by aspects of metabolism. Roizen et al. (37) found that the serum concentration of 25OHD, but not vitamin D, was decreased in mice fed a high fat diet to induce obesity compared with normal weight mice. Moreover, mRNA and protein levels of CYP2R1 were decreased in these obese mice. The expression of other 25-hydroxylases (CYP27A1, CYP3A) or the catabolizing enzyme CYP24A1 was not altered. Aatsinki et al (38) examined the effect of high fat diet induced obesity, fasting, and type 2 diabetes as well as streptozotocin induced (type 1) diabetes on 25OHD levels in mice. All these metabolic manipulations decreased the hepatic mRNA and protein concentration of CYP2R1. These authors then demonstrated that the decrease in CYP2R1 was mediated by PPARγ-coactivator-1α (PGC1α), a key metabolic regulator increased by fasting or diabetes. They then showed that the control of CYP2R1 gene expression by PGC1α involved another transcriptional regulator, estrogen-related receptor α (ERRα), which also binds to other nuclear receptors such as VDR and the glucocorticoid receptor (GR). Consistent with this is that dexamethasone, a ligand for GR, decreased hepatic CYP2R1 mRNA and protein concentrations by a mechanism mediated by increased PGC1α.
Renal Production of 1,25(OH)2D
1,25(OH)2D is the most potent metabolite of vitamin D, and mediates most of its hormonal actions. 1,25(OH)2D is produced from 25OHD by the enzyme 25OHD-1α hydroxylase (CYP27B1). The cloning of CYP27B1 by four independent groups (40-43) ended a long effort to determine the structure of this critical enzyme in vitamin D metabolism. Mutations in this gene are responsible for the rare autosomal disease of pseudovitamin D deficiency rickets (40,42,44,45). An animal model in which the gene is knocked out by homologous recombination reproduces the clinical features of this disease including retarded growth, rickets, hypocalcemia, hyperparathyroidism, and undetectable 1,25(OH)2D (46). Unlike Vdr null mice and VDR mutations in humans, alopecia is not part of this phenotype.
CYP27B1 is a mitochondrial mixed function oxidase with significant homology to other mitochondrial steroid hydroxylases including CYP27A1 (39%), CYP24A1 (30%), CYP11A1 (32%), and CYP11β (33%) (40). However, within the heme-binding domain the homology is much greater with 73% and 65% sequence identity with CYP27A1 and CYP24A1 (40). These mitochondrial P450 enzymes are located in the inner membrane of the mitochondrion, and serve as the terminal acceptor for electrons transferred from NADPH through ferrodoxin reductase and ferrodoxin. Expression of CYP27B1 is highest in epidermal keratinocytes (40), cells that previously had been shown to contain high levels of this enzymatic activity (47). However, the kidney also expresses this enzyme in the renal tubules as do the brain, placenta, testes, intestine, lung, breast, macrophages, lymphocytes, parathyroid gland, osteoblasts and chondrocytes (40,48-51). That said, the kidney is generally considered the major source of circulating levels of 1,25(OH)2D, with the extrarenal CYP27B1 activities providing for local needs under normal circumstances. However, extrarenal sources can lead to increased 1,25(OH)2D and calcium levels in some pathologic conditions to be discussed subsequently.
The principal regulators of CYP27B1 activity in the kidney are parathyroid hormone (PTH), FGF23, calcium, phosphate, and 1,25(OH)2D. Extrarenal production tends to be stimulated by cytokines such as IFN-gamma and TNF-α more effectively than PTH (52) and may be less inhibited by calcium, phosphate, and 1,25(OH)2D depending on the tissue. Administration of PTH in vivo (53) or in vitro (54,55) stimulates renal production of 1,25(OH)2D. This action of PTH can be mimicked by cAMP (53,55) and forskolin (56,57) indicating that at least part of the effect of PTH is mediated via its activation of adenylate cyclase. However, PTH activation of protein kinase C (PKC) also appears to be involved in that concentrations of PTH sufficient to stimulate PKC activation and 1,25(OH)2D production are below that required to increase cAMP levels (58). Furthermore, synthetic fragments of PTH lacking the ability to activate adenylate cyclase but which stimulate PKC activity were found to increase 1,25(OH)2D production (59). Direct activation of PKC with phorbol esters results in increased 1,25(OH)2D production. Although the promoter of CYP27B1 contains several AP-1 (PKC activated) and cAMP response elements, it is not yet clear how PTH regulates CYP27B1 gene expression (60). However, several mechanisms have been proposed. In one study the nuclear receptor 4A2 acting through a C/EBP consensus element appears to be involved (61). Another mechanism involves VDIR that is proposed to bind to a negative VDRE in the CYP27B1 promoter. When PKA is activated by PTH VDIR is phosphorylated and recruits the p300 complex with HAT activity, inducing gene transcription (62). Calcium modulates the ability of PTH to increase 1,25(OH)2D production. Calcium by itself can decrease CYP27B1activity (63,64) and block the stimulation by PTH (65). Given in vivo, calcium can exert its effect in part by reducing PTH secretion, but this does not explain its direct actions in vitro or its effects in parathyroidectomized or PTH infused animals. Phosphate deprivation can stimulate CYP27B1 activity in vivo (66,67) and in vitro (68). The in vivo actions of phosphate deprivation can be blocked by hypophysectomy (69,70) and partially restored by growth hormone (GH) (70,71) and insulin-like growth factor (IGF-I) (72). However, like PTH, the exact mechanism by which GH and/or IGF-I mediates the effects of phosphate on CYP27B1 expression remains unclear. More recently FGF23 has been shown to inhibit CYP27B1 activity in vivo and in vitro (73). FGF23 has been implicated as at least one of the factors responsible for impaired phosphate reabsorption and 1,25(OH)2D production in conditions such as X-linked and autosomal dominant hypophosphatemic rickets and oncogenic osteomalacia (74,75). FGF23 acts through FGF receptors 1 and 3 in conjunction with the coreceptor Klotho, but the mechanism by which FGF23 regulates CYP27B1 remains obscure. High phosphate stimulates FGF23 production from bone, and this is likely the major mechanism by which phosphate leads to decreased CYP27B1 activity (76). 1,25(OH)2D administration leads to reduction in CYP27B1 activity. In the kidney Meyer et al. (77) identified a region in the Cyp27b1 gene that when deleted blocked 1,25(OH)2D production. However, in other tissues no vitamin D response element has been identified in the promoter of the 1α-hydroxylase gene (60). In keratinocytes, 1,25(OH)2D has little or no effect on CYP27B1 mRNA and protein levels when given in vitro. When 24-hydroxylase activity is blocked, 1,25(OH)2D administration fails to reduce the levels of 1,25(OH)2D produced (78,79). Thus, the apparent feedback regulation of CYP27B1 activity by 1,25(OH)2D in most tissues, with the possible exception of the kidney, appears to be due to its stimulation of CYP24A1 and subsequent catabolism, not to a direct effect on CYP27B1 expression or activity. Moreover, 1,25(OH)2D stimulates FGF23 production and inhibits PTH production. Both actions will decrease, indirectly, the ability of 1,25(OH)2D to inhibit its own production (76). Thus, renal and extrarenal regulation of CYP27B1 by 1,25(OH)2D may differ.
Renal Production of 24,25(OH)2D
The kidney is also the major producer of a second important metabolite of 25OHD, namely 24,25(OH)2D, and the enzyme responsible is 25OHD-24 hydroxylase (CYP24A1) [75]. CYP24A1 and CYP27B1 are homologous enzymes that coexist in the mitochondria of tissues where both are found, such as the kidney tubule. However, there genes are located on different chromosomes (chromosome 20q13 and chromosome 12q14 for CYP24A1 and CYP27B1, respectively, in humans). They share the same ferrodoxin and ferrodoxin reductase components. While CYP27B1 activates the parent molecule, 25OHD, CYP24A1 initiates a series of catabolic steps that lead to its inactivation. However, in some tissues 24,25(OH)2D has been shown to have biologic effects different from 1,25(OH)2D as will be described subsequently. CYP24A1 24-hydroxylates both 25OHD and 1,25(OH)2D. The 24-hydroxylation is then followed by oxidation of 24OH to a 24-keto group, 23-hydroxylation, cleavage between C23-24, and the eventual production of calcitroic acid, a metabolite with no biologic activity. CYP24A1 also has 23-hydroxylase activity, initiating steps that lead to 23/26 lactone formation. Different species have CYP24A1s that differ in their preference for the 24-hydroxylation vs 23-hydroxylation pathway. The human enzyme follows the 24-hydroxylation pathway. Analogs with differences in their side chain are also likely to differ in the pathway utilized. CYP24A1 catalyzes all the steps in this catabolic pathway (81) (82). Although CYP24A1 is highly expressed in the kidney tubule, its tissue distribution is quite broad. In general, CYP24A1 can be found wherever the VDR is found. The affinity for 1,25(OH)2D is higher than that for 25OHD, making this enzyme an efficient means for eliminating 1,25(OH)2D. Thus, CYP24A1 is likely to play the important role of protecting the body against excess 1,25(OH)2D. Indeed, inactivating mutations in CYP24A1 have been found to underlie the disease idiopathic infantile hypercalcemia (83), manifesting as the name suggests with elevated serum calcium and 1,25(OH)2D levels. These individuals may present for the first time as adults, often in the context of increased 1,25(OH)2D production as in pregnancy (84). An animal model in which CYP24A1 has been knocked out likewise showed very high levels of 1,25(OH)2D when treated with vitamin D and impaired mineralization of intramembranous bone (85). The skeletal abnormalities could be corrected by crossing this mouse to one lacking the VDR suggesting that excess 1,25(OH)2D (which acts through the VDR) rather than deficient 24,25(OH)2D (which does not) is to blame (85).
The regulation of CYP24A1 in the kidney is almost the mirror image of that of CYP27B1. PTH and 1,25(OH)2D are the dominant regulators, but calcium, phosphate, insulin, FGF23, IGF-I, GH, and sex steroids may also play a role. 1,25(OH)2D induces CYP24A1. The promoter of CYP24A1 has two vitamin D response elements (VDREs) critical for this induction (86-88). Protein kinase C activation as by phorbol esters enhances this induction by 1,25(OH)2D (89). An AP-1 site is found adjacent to the proximal VDRE, but mutation of this site does not appear to block phorbol ester enhancement of CYP24A1 induction by 1,25(OH)2D (90). PTH, on the other hand, inhibits the expression of CYP24A1 in the kidney (91). This action can be reproduced with cAMP (92) and forskolin (56) indicating the role of PTH activated adenylate cyclase (93). PTH has no effect on intestinal CYP24A1, most likely because the intestine does not have PTH receptors. Surprisingly, however, PTH is synergistic with 1,25(OH)2D in stimulating CYP24A1 expression and activity in bone cells which do have PTH receptors, again through a cAMP mediated mechanism (94). This synergism is further potentiated by the addition of insulin (95) (96). FGF23 also induces CYP24A1 expression (97). Surprisingly this requires the VDR (97), since FGF23 also inhibits 1,25(OH)2D production and so would be expected to reduce CYP24A1 via a 1,25(OH)2D/VDR mechanism. Restriction in dietary phosphate reduces CYP24A1 expression consistent with a decrease in FGF23, but also in a manner blocked by hypophysectomy (98). GH and IGF-I can reduce CYP24A1 expression in hypophysectomized animals, suggesting that the phosphate effect on CYP24A1 like its opposing effect on CYP27B1, is mediated by GH and IGF-I (98) as well as FGF23. The region(s) of the CYP24A1 promoter mediating these actions of PTH and FGF23 as well as 1,25(OH)2D have recently been mapped (96). Similar to that for CYP27B1 this regulation differs in different cell types. Thus, although different regulators tend to have opposite effects on CYP24A1 and CYP27B1 expression the molecular mechanisms by which the regulation occurs also differ for each enzyme.
TRANSPORT IN BLOOD
The vitamin D metabolites are transported in blood bound primarily to vitamin D binding protein (DBP) (85-88%) and albumin (12-15%) (99-101). DBP concentrations are normally 4-8µM, well above the concentrations of the vitamin D metabolites, such that DBP is only about 2% saturated. DBP has high affinity for the vitamin D metabolites (Ka=5x108M-1 for 25OHD and 24,25(OH)2D, 4x107M-1 for 1,25(OH)2D and vitamin D), such that under normal circumstances only approximately 0.03% 25OHD and 24,25(OH)2D and 0.4% 1,25(OH)2D are free (100-102). Conditions such as liver disease and nephrotic syndrome resulting in reduced DBP and albumin levels will lead to a reduction in total 25OHD and 1,25(OH)2D levels without necessarily affecting the free concentrations (103) (figure 3). Similarly, DBP levels are reduced during acute illness, potentially obscuring the interpretation of total 25OHD levels (104). Earlier studies with a monoclonal antibody to measure DBP levels suggested a decreased level in African Americans consistent with their lower total 25OHD levels, but these results were not confirmed using polyvalent antibody-based assays (105). Vitamin D intoxication can increase the degree of saturation sufficiently to increase the free concentrations of 1,25(OH)2D and so cause hypercalcemia without necessarily raising the total concentrations (106).
The vitamin D metabolites bound to DBP are in general not available to most cells. Thus, the free or unbound concentration is that which is critical for cellular uptake as postulated by the free hormone hypothesis. Support for the concept that the role of DBP is to provide a reservoir for the vitamin D metabolites but that it is the free concentration that enters cells and exerts biologic function comes from studies in mice in which DBP has been deleted and in humans in which the gene is mutated. In DBP knockout mice the vitamin D metabolites are presumably all free and/or bioavailable. These mice do not show evidence of vitamin D deficiency unless placed on a vitamin D deficient diet despite having very low levels of serum 25OHD and 1,25(OH)2D (107). Tissue levels of 1,25(OH)2D were found to be normal in the DBP knockout mice as were markers of vitamin D action such as expression of intestinal TRPV6, calbindin 9k, PMCA1b, and renal TRPV5 (108). Recently a family in which a large deletion of the coding portion of the DBP gene (and adjacent NPFFR2 gene) has been reported (109). The proband had normal calcium, phosphate and PTH levels with vitamin D supplementation despite very low levels of 25OHD, 24,25(OH)2D, and 1,25(OH)2D that were not responsive to massive doses of vitamin D (oral or parenteral). The free 25OHD was nearly normal. The carrier sibling had vitamin D metabolite levels between those of the proband and the normal sibling. Thus, both the studies in DBP null mice and humans support the free hormone hypothesis while also supporting the role of DBP as a circulating reservoir for the vitamin D metabolites. Therefore, there is currently a debate as to whether the free concentration of 25OHD, for example, is a better indicator of vitamin D nutritional status than total 25OHD, given that DBP levels, and hence total 25OHD levels, can be influenced by liver disease, nephrotic syndrome, pregnancy, and inflammatory states (110,111). However, certain tissues such as the kidney, placenta, and parathyroid gland express the megalin/cubilin complex which is able to transport vitamin D metabolites bound to DBP into the cell. This is critical for preventing renal losses of the vitamin metabolites (112) and may be important for vitamin D metabolite transport into the fetus and regulation of PTH secretion. Indeed, mice lacking the megalin/cubilin complex have poor survival with evidence of osteomalacia indicating its role in vitamin D transport into critical cells involved with vitamin D signaling
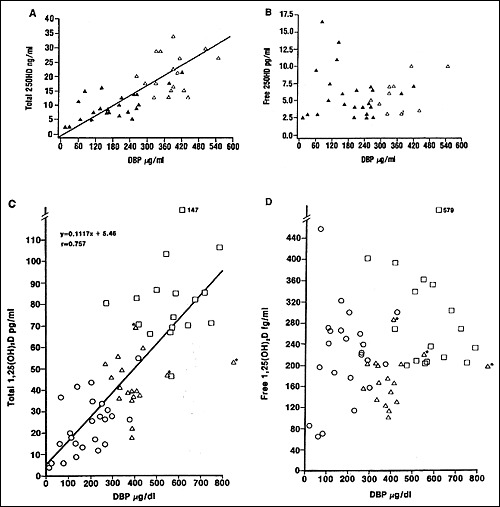
Figure 3.
Correlation of total 25OHD (A) and 1,25(OH)2D (C) levels to DBP; lack of correlation of free 25OHD (B) and 1,25(OH)2D (D) levels to DBP. Data from normal subjects (open triangles), subjects with liver disease (closed triangles, open circles), subjects on oral contraceptives (open triangles*), and pregnant women (open squares) are included. These data demonstrate the dependence of total 25OHD and 1,25(OH)2D concentrations on DBP levels which are reduced by liver disease. However, the free concentrations of 25OHD and 1,25(OH)2D are normal in most patients with liver disease. Reprinted with permission from the American Society for Clinical Investigation.
DBP was originally known as group specific component (Gc-globulin) before its properties as a vitamin D transport protein became known. It has three common polymorphisms which are useful in population genetics. These alleles have somewhat different affinities for the vitamin D metabolites (113), but which do not appear to alter its function. DBP is a 58kDa protein with 458 amino acids that is homologous to albumin and α-fetoprotein (αFP) (40% homology at the nucleotide level, 23% at the amino acid level) (114). These three genes cluster on chromosome 4q11-13 (115). DBP, like albumin and αFP, is made primarily but not exclusively in the liver-other sites include the kidney, testes, and fat. DBP like other steroid hormone binding proteins is increased by oral (not transdermal) estrogens and pregnancy (100). In vitro, glucocorticoids and cytokines such as EGF, IL-6 and TGF-β have been shown to increase (glucocorticoids, EGF, IL-6) or decrease (TGF-β) DBP production (116).
Although transport of the vitamin D metabolites may be the major function for DBP, it has other properties. DBP has high affinity for actin, and may serve as a scavenger for actin released into the blood during cell death (117). DBP has also been shown to activate macrophages (118) and osteoclasts (119). However, in a mouse rendered deficient in DBP by homologous recombination (knock out) no obvious abnormality was observed except for increased turnover in vitamin D and increased susceptibility to osteomalacia on a vitamin D deficient diet (120). Evidence for osteopetrosis (indicating failure of osteoclast function) was not found.
MECHANISM OF ACTION
The hormonal form of vitamin D, 1,25(OH)2D, is the ligand for a transcription factor, the vitamin D receptor (VDR). Most if not all effects of 1,25(OH)2D are mediated by VDR acting primarily by regulating the expression of genes whose promoters contain specific DNA sequences known as vitamin D response elements (VDREs). There are thousands of VDREs throughout the gene, often thousands of base pairs away from the coding portion of the gene regulated. However, some actions of 1,25(OH)2D are more immediate, and may be mediated by a membrane bound vitamin D receptor that has been less well characterized than the nuclear VDR or by the VDR acting outside of the nucleus. On the other hand, some actions of VDR do not require its ligand 1,25(OH)2D. Our understanding of the mechanism by which VDR regulates gene expression has increased enormously over the past few years.
VDR and Transcriptional Regulation
The VDR was discovered in 1969 (121) (although only as a binding protein for an as yet unknown vitamin D metabolite subsequently identified as 1,25(OH)2D), and was eventually cloned and sequenced in 1987 (122,123). Inactivating mutations in the VDR result in hereditary vitamin D resistant rickets (HVDRR) (124). Animal models in which the VDR has been knocked out (125) (126) have the full phenotype of severe vitamin D deficiency indicating that the VDR is the major mediator of vitamin D action. The one major difference is the alopecia seen in HVDRR and VDR knockout animals, a feature not associated with vitamin D deficiency, suggesting that the VDR may have functions independent of 1,25(OH)2D at least in hair follicle cycling. The VDR is a member of a large family of proteins (over 150 members) that includes the receptors for the steroid hormones, thyroid hormone, vitamin A family of metabolites (retinoids), and a variety of cholesterol metabolites, bile acids, isoprenoids, fatty acids and eicosanoids. A large number of family members have no known ligands, and are called orphan receptors. VDR is widely, although not universally, distributed throughout the different tissues of the body (127). Many of these tissues were not originally considered target tissues for 1,25(OH)2D. The discovery of VDR in these tissues along with the demonstration that 1,25(OH)2D altered function of these tissues has markedly increased our appreciation of the protean effects of 1,25(OH)2D.
The VDR is a molecule of approximately 50-60kDa depending on species. The basic structure is shown in figure 4. The VDR is unusual in that it has a very short N-terminal domain before the DNA binding domain when compared to other nuclear hormone receptors. The human VDR has two potential start sites. A common polymorphism (Fok 1) alters the first ATG start site to ACG. Individuals with this polymorphism begin translation three codons downstream such that in these individuals the VDR is three amino acids shorter (424 aas vs 427 aas). This polymorphism has been correlated with reduced bone density suggesting it is of functional importance (128). The most conserved domain in VDR from different species and among the nuclear hormone receptors in general is the DNA binding domain. This domain is comprised of two zinc fingers. The name derives from the cysteines within this stretch of amino acids that form tetrahedral complexes with zinc in a manner which creates a loop or finger of amino acids with the zinc complex at its base. The proximal (N-terminal) zinc finger confers specificity for DNA binding to the VDREs while the second zinc finger and the region following provide at least one of the sites for heterodimerization of the VDR to the retinoid X receptor (RXR). The second half of the molecule is the ligand binding domain, the region responsible for binding 1,25(OH)2D, but which also contains regions necessary for heterodimerization to RXR. At the C-terminal end is the major activation domain, AF-2, which is critical for the binding to coactivators such as those in the steroid receptor coactivator (SRC) and vitamin D receptor interacting protein (DRIP) or Mediator families (129). In mutation studies of the homologous thyroid receptor, corepressors were found to bind in overlapping regions with coactivators in helices 3 and 5, a region blocked by helix 12 (the terminal portion of the AF2 domain) in the presence of ligand (130). Deletion of helix 12 promoted corepressor binding while preventing that of coactivators (130).
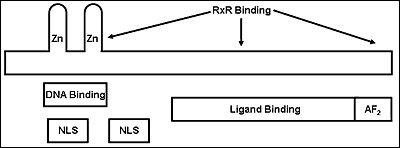
Figure 4.
Model of the vitamin D receptor (VDR). The N terminal region is short relative to other steroid hormone receptors. This region is followed by two zinc fingers which constitute the principal DNA binding domain. Nuclear localization signals (NLS) are found within and just C-terminal to the DNA binding domain. The ligand binding domain makes up the bulk of the C-terminal half of the molecule, with the AF2 domain comprising the most C-terminal region. The AF2 domain is largely responsible for binding to co-activators such as the SRC family and DRIP (Mediator) in the presence of ligand. Regions on the second zinc finger and within the ligand binding domain facilitate heterodimerization with RXR. Corepressor binding is less well characterized but appears to overlap that of coactivators in helices 3 and 5, a region blocked by helix 12 in the presence of ligand.
The ligand binding domain (LBD) for VDR has been crystallized and its structure solved (131). More recently the structure of the VDR/RXR heterodimer has been analyzed by high resolution cryoelectron microscopy (132). These studies show that the VDR has a high degree of structural homology to other nuclear hormone receptors. It is comprised of 12 helices joined primarily by beta sheets. The 1,25(OH)2D is buried deep in the ligand binding pocket and covered by helix 12 (the terminal portion of the AF-2 domain). Assuming analogy with the unliganded LBD of RXRα and the ligand bound LBD of RARγ (133), the binding of 1,25(OH)2D to the VDR triggers a substantial movement of helix 12 from an open position to a closed position, covering the ligand binding pocket and putting helix 12 in position with critical residues from helices 3, 4, and 5 to bind coactivators. Coactivator complexes bridge the gap from the VDRE to the transcription machinery at the transcription start site (figure 5) (134).

Figure 5.
1,25(OH)2D-initiated gene transcription. 1,25(OH)2D enters the target cell and binds to its receptor, VDR. The VDR then heterodimerizes with the retinoid X receptor (RXR). This increases the affinity of the VDR/RXR complex for the vitamin D response element (VDRE), a specific sequence of nucleotides in the promoter region of the vitamin D responsive gene. Binding of the VDR/RXR complex to the VDRE attracts a complex of proteins termed coactivators to the VDR/RXR complex. The DRIP (Mediator) coactivator complex spans the gap between the VDRE and RNA polymerase II and other proteins in the initiation complex centered at or around the TATA box (or other transcription regulatory elements). SRC coactivators recruit histone acetyl transferases (HAT) to the gene promoting the opening up of its structure to enable the transcription machinery to work. Transcription of the gene is initiated to produce the corresponding mRNA, which leaves the nucleus to be translated to the corresponding protein.
Nuclear hormone receptors including the VDR are further regulated by protein complexes that can be activators or repressors (135). The role of corepressors in VDR function has been demonstrated (136) but is less well studied than the role of coactivators. One such corepressor, hairless, is found in the skin and may regulate 1,25(OH)2D mediated epidermal proliferation and differentiation as well as ligand independent VDR regulation of hair follicle cycling (137-139). The coactivators, which are essential for VDR function, form two distinct complexes, the interaction of which remains unclear (129). The SRC family has three members, SRC 1-3, all of which can bind to the VDR in the presence of ligand (1,25(OH)2D) (140). These coactivators recruit additional coactivators such as CBP/p300 and p/CAF that have histone acetyl transferase activity (HAT), an enzyme that by acetylation of lysines within specific histones appears to help unravel the chromatin allowing the transcriptional machinery to do its job. The domain in these molecules critical for binding to the VDR and other nuclear hormone receptors is called the NR box, and has as its central motif LxxLL where L stands for leucine and x for any amino acid. Each SRC family member contains three well conserved NR boxes in the region critical for nuclear hormone receptor binding. The DRIP (Mediator) complex is comprised of 15 or so proteins several of which contain LxxLL motifs (141). However, DRIP205 (Mediator 1) is the protein critical for binding the complex to VDR. It contains 2 NR boxes. Different NR boxes in these coactivators show specificity for different nuclear hormone receptors (142). Unlike the SRC complex, the DRIP complex does not have HAT activity (129). Rather the DRIP complex spans the gene from the VDRE to the transcription start site linking directly with RNA polymerase II and its associated transcription factors. DRIP and SRC appear to compete for binding to the VDR. In keratinocytes DRIP binds preferentially to the VDR in undifferentiated cells, whereas SRC 2 and 3 bind in the more differentiated cells in which DRIP levels have declined (143). Thus in these cells DRIP appears to regulate the early stages of 1,25(OH)2D induced differentiation, whereas SRC may be more important in the later stages, although overlap in gene specificity is also observed (144,145) (146). These coregulators are not specific for VDR, but interact with a large number of other transcription factors. The DRIP (Mediator) complex can mark regions in the genome containing large numbers of sites for transcription factors including VDREs. These sites are known as super enhancers often regulating genes involved with cell fate determination (147). Recently, SMAD 3, a transcription factor in the TGF-β pathway, has been found to complex with the SRC family members and the VDR, enhancing the coactivation process (148). Phosphorylation of the VDR may also control VDR function (149). Furthermore, VDR has been shown to suppress β-catenin transcriptional activity (150), whereas β-catenin enhances that of VDR (151). Thus, control of VDR activity may involve crosstalk between signaling pathways originating in receptors at the plasma membrane as well as within the nucleus.
VDR acts in concert with other nuclear hormone receptors, in particular RXR (152). Unlike VDR, there are three forms of RXR--α, β, γ--and all three are capable of binding to VDR with no obvious differences in terms of functional effect. RXR and VDR form heterodimers that optimize their affinity for the vitamin D response elements (VDREs) in the genes being regulated. RXR appears to be responsible for keeping VDR in the nucleus in the absence of ligand (153). VDR may also partner with other receptors including the thyroid receptor (TR) and the retinoic acid receptor (RAR) (154,155), but these are the exceptions, whereas RXR is the rule. The VDR/RXR heterodimers bind to VDREs, which typically are comprised of two half sites each with six nucleotides separated by three nucleotides of nonspecific type; this type of VDRE is known as a DR3 (direct repeats with three nucleotide spacing). RXR binds to the upstream half site, while VDR binds to the downstream site (156). However, a wide range of VDRE configurations have been found at nearly any location within a gene (5’, 3’, introns) (157). Moreover, different tissues differ as to which VDREs actively bind VDR (158). 1,25(OH)2D is required for high affinity binding and activation, but the RXR ligand, 9-cis retinoic acid, may either inhibit (159) or activate (160) 1,25(OH)2D stimulation of gene transcription. A DR6 has been identified in the phospholipase C-γ1 gene that recognizes VDR/RAR heterodimers (154), and a DR4 has been found in the mouse calbindin 28k gene (161). Inverted palinodromes with 7 to 12 bases between half sites have also been found (151). Furthermore, the half sites of the various known VDREs show remarkable degeneracy (table 1). The G in the second position of each site appears to be the only nearly invariant nucleotide. 1,25(OH)2D can also inhibit gene transcription through its VDR. This may occur by direct binding of the VDR to negative VDREs that in the PTH and PTHrP genes are remarkably similar in sequence to positive VDREs of other genes (162,163). However, inhibition may also be indirect. For example, 1,25(OH)2D inhibits IL-2 production by blocking the NFATp/AP-1 complex of transcription factors from activating this gene (164) through a mechanism not yet clear. Similarly, 1,25(OH)2D inhibits CYP27B1 in at least one renal cell line by an indirect mechanism involving VDR binding to VDIR (62,80). Thus, a variety of factors including the flanking sequences of the genes around the VDREs and tissue specific factors play a large role in dictating the ability of 1,25(OH)2D to regulate gene expression.
Non-Genomic Actions
A variety of hormones that serve as ligands for nuclear hormone receptors also exert biologic effects that do not appear to require gene regulation and may work through membrane receptors rather their cognate nuclear hormone receptors. Examples include estrogen (165), progesterone (166), testosterone (167), corticosteroids (168), and thyroid hormone (169). 1,25(OH)2D has also been shown to have rapid effects on selected cells that are not likely to involve gene regulation and that appear to be mediated by a different, probably membrane receptor. A model for such effects is shown in figure 6. Similar to other steroid hormones, 1,25(OH)2D has been shown to regulate calcium and chloride channel activity, protein kinase C activation and distribution, and phospholipase C activity in a number of cells including osteoblasts (170), liver (171), muscle (172), and intestine (173,174). These rapid effects of 1,25(OH)2D have been most extensively studied in the intestine. Norman's laboratory coined the term transcaltachia to describe the rapid onset of calcium flux across the intestine of a vitamin D replete chick perfused with 1,25(OH)2D (175). This increased flux could not be blocked with actinomycin D pretreatment (176), but was blocked by voltage gated L type channel inhibitors (177) and protein kinase C inhibitors (178). These animals had to be vitamin D replete and contain the VDR, indicating that the basic machinery for calcium transport was intact. On the other hand L type channel activators such as BAY K-8644 (179) and protein kinase C activators such as phorbol esters (177) could activate transcaltachia similar to 1,25(OH)2D.
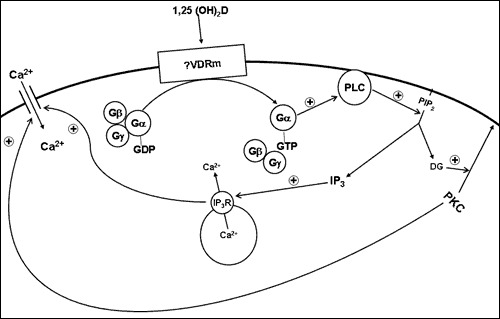
Figure 6.
Model for the non-genomic actions of 1,25(OH)2D. 1,25(OH)2D binds to a putative membrane receptor. This leads to activation of a G protein (GTP displacement of GDP and dissociation of the β and γ subunits from the now active α subunit). Gα -GTP activates phospholipase C (PLC) (β or γ) to hydrolyze phosphatidyl inositol bis phosphate (PIP2) to inositol tris phosphate (IP3) and diacyl glycerol (DG). IP3 releases calcium from intracellular stores via the IP3 receptor in the endoplasmic reticulum; DG activates protein kinase C (PKC). Both calcium and PKC may regulate the influx of calcium across the plasma membrane through various calcium channels including L-type calcium channels.
A putative membrane receptor for 1,25(OH)2D (1,25(OH)2D membrane associated rapid response steroid binding protein (1,25D-MARRSBP) also known as ERp57) has been purified from the intestine (180) and subsequently cloned and sequenced (181). Its size is approximately 66kDa. Antibodies have been made against this putative receptor (182). These antibodies block the ability of 1,25(OH)2D to stimulate calcium uptake by isolated chick intestinal cells (183) and to stimulate protein kinase C activity in resting zone chondrocytes while inhibiting proliferation of both resting zone and proliferating zone chondrocytes (182). Analog studies also support the existence of a separate membrane receptor for 1,25(OH)2D. Because of the breaking of the B ring during vitamin D3 production from 7-dehydrocholesterol, the A ring can assume a conformation similar to the parent cholesterol molecule (6-s-cis) (shown as previtamin D3 in figure 1) or the more commonly depicted 6-s-trans form in which the A ring rotates away from the rest of the molecule (shown as vitamin D3 in figure 1). Analogs of 1,25(OH)2D can be produced which favor the 6-s-cis conformation or the 6-s-trans conformation. 1,25(OH)2-d5-previtamin D3 is one such analog locked into the 6-s-cis conformation. This analog has only weak activity with respect to VDR binding or transcriptional activation but is fully effective in terms of stimulating transcaltachia and calcium uptake by osteosarcoma cells when compared to 1,25(OH)2D (184). 6-s-trans analogs are not effective. However, some of these rapid actions of 1,25(OH)2D are not found in cells from VDR null mice suggesting that the VDR may be required for the expression and/or function of the membrane receptor or be the membrane receptor. In other cells both 1,25D-MARRSBP and VDR appear to be required for these rapid effects of 1,25(OH)2D (185,186).
The model (figure 6) emerging from these studies is that 1,25(OH)2D interacts with a membrane receptor to activate phospholipase C possibly through a G protein coupled process. Phospholipase C then hydrolyzes phosphatidyl inositol bis phosphate (PIP2) in the membrane releasing inositol tris phosphate (IP3) and diacyl glycerol (DG). These second messengers may then activate both the intracellular release of calcium from intracellular stores via the IP3 receptor and protein kinase C, either one or both of which could stimulate calcium channel activity leading to a further rise in intracellular calcium levels. In the intestine and kidney, the increased flux of calcium across the brush border membrane is then transported out of the cell at the basolateral membrane, completing transcellular transport. In other cells the increased calcium would need to be removed by other mechanisms after the signal conveyed by the rise in calcium is no longer required. Much work remains to prove this model including the physiologic requirement for a unique membrane receptor.
TARGET TISSUE RESPONSES: CALCIUM REGULATING ORGANS
Intestine
Intestinal calcium absorption, in particular the active component of transcellular calcium absorption, is one of the oldest and best known actions of vitamin D having been first described in vitro by Schachter and Rosen (187) in 1959 and in vivo by Wasserman et al. (188) in 1961. Absorption of calcium from the luminal contents of the intestine involves both transcellular and paracellular pathways. The transcellular pathway dominates in the duodenum and cecum, and this is the pathway primarily regulated by 1,25 dihydroxyvitamin D (1,25(OH)2D) (189), although elements of the paracellular pathway such as the claudins 2 and 12 are likewise regulated by 1,25(OH)2D (reviews in (190,191). Figure 7 shows a model of our current understanding of how this process is regulated by 1,25(OH)2D. Calcium entry across the brush border membrane (BBM) occurs down a steep electrical-chemical gradient and requires no input of energy. Removal of calcium at the basolateral membrane must work against this gradient, and energy is required. This is achieved by the CaATPase (PMCA1b), an enzyme induced by 1,25(OH)2D in the intestine. Calcium movement through the cell occurs with minimal elevation of the intracellular free calcium concentration (192) by packaging the calcium in calbindin containing vesicles (193-195) that form in the terminal web following 1,25(OH)2D administration.
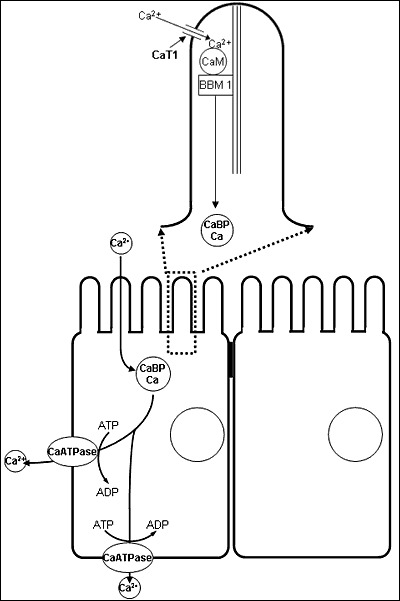
Figure 7.
Model of intestinal calcium transport. Calcium enters the microvillus of the intestinal epithelial cell through TRPV6 (previously known as CaT1) calcium channel. Within the microvillus calcium is bound to calmodulin (CaM) which is itself bound to brush border myosin I (BBMI). BBMI may facilitate the movement of the calcium/CaM complex into the terminal web where the calcium is picked up by calbindin (CaBP) and transported through the cytoplasm in endocytic vesicles. At the basolateral membrane the calcium is pumped out of the cell by the Ca-ATPase (PMCA1b). 1,25(OH)2D enhances intestinal calcium transport by inducing TRPV6, CaBP, and PMCAb as well as increasing the amount of CaM bound to BBMI in the brush border.
1,25(OH)2D regulates transcellular calcium transport using a combination of genomic and nongenomic actions. The first step, calcium entry across the BBM, is accompanied by changes in the lipid composition of the membrane including an increase in linoleic and arachidonic acid (196,197) and an increase in the phosphatidylcholine:phosphatidylethanolamine ratio (198). These changes are associated with increased membrane fluidity (197), which we have shown results in increased calcium flux (199). The changes in lipid composition occur within hours after 1,25(OH)2D administration and are not blocked by pretreatment with cycloheximide (198). In addition, an epithelial specific calcium channel, TRPV6, is expressed in the intestinal epithelium (200). This channel has a high degree of homology to TRPV5, a channel originally identified in the kidney (201,202). The tissue distributions of these channels are overlapping and can be found in other tissues, but TRPV6 appears to be the main form in the intestine (203,204). TRPV6 mRNA levels in the intestine of vitamin D deficient mice are markedly increased by 1,25(OH)2D, although similar changes are not found in the kidney (205). Mice null for TRPV6 have decreased intestinal calcium transport (206).
Calcium entering the brush border must then be moved into and through the cytoplasm without disrupting the function of the cell. Electron microscopic observations indicate that in the vitamin D deficient animal, calcium accumulates along the inner surface of the plasma membrane of the microvilli (207,208). Following vitamin D or 1,25(OH)2D administration calcium leaves the microvilli and subsequently can be found in mitochondria and vesicles within the terminal web (193,194,207,208). The vesicles appear to shuttle the calcium to the lateral membrane where it is pumped out of the cell by the basolateral CaATPase, PMCA1b. These morphologic observations have been confirmed by direct measurements of calcium using x-ray microanalysis that demonstrate equivalent amounts of calcium within the microvilli of D deficient and 1,25(OH)2D treated animals but much higher amounts of calcium in the mitochondria and vesicles of the 1,25(OH)2D treated animals (194,209). Such data suggest that 1,25(OH)2D controls calcium entry into the cell primarily by regulating its removal from the microvillus and accumulation by subcellular organelles in the terminal web, although flux through calcium channels in the membrane such as TRPV6 also plays a major role.
The ability of 1,25(OH)2D to stimulate calcium entry into and transport from the microvillus does not require new protein synthesis (193,198,210). Cycloheximide does not block the ability of 1,25(OH)2D to increase the capacity of brush border membrane vesicles (BBMV) to accumulate calcium, although it does block the increase in alkaline phosphatase in the same BBMV [193]. Likewise, cycloheximide does not block the increase in mitochondrial calcium following 1,25(OH)2D administration, although it blocks the rise in calbindin and prevents the normal vesicular transport of calcium through the cytosol (193,211). Thus, nongenomic actions underlie at least some of these first steps in 1,25(OH)2D stimulated intestinal calcium transport within the microvillus, although the changes take hours, not minutes, to observe. The exact role for these nongenomic effects on calcium influx relative to the role of TRPV6 remains to be elucidated.
Calmodulin is the major calcium binding protein in the microvillus (212). Its concentration in the microvillus is increased by 1,25(OH)2D; no new calmodulin synthesis is required or observed after 1,25(OH)2D administration (213). Calmodulin is likely to play a major role in calcium transport within the microvillus, and inhibitors of calmodulin block 1,25(OH)2D stimulated calcium uptake by BBMV (214). Within the microvillus calmodulin is bound to a 110kD protein, myosin 1A (myo1A)) (previously referred to as brush border myosin 1). 1,25(OH)2D increases the binding of calmodulin to myo1A in brush border membrane preparations (213), although binding of calmodulin to the myo1A attached to the actin core following detergent extraction of the membrane appears to be reduced (215). The calmodulin/myo1A complex appears late in the development of the brush border, and is found in highest concentration in the same cells of the villus which have the highest capacity for calcium transport (216). Myo1A is located primarily in the microvillus of the mature intestinal epithelial cell, although small amounts have been detected associated with vesicles in the terminal web (217). Thus, the calmodulin/myo1A complex may be responsible for moving calcium out of the microvillus. Its exact role in calcium transport is unclear in that mice null for myo1A do not show reduced intestinal calcium transport(218)). Calbindin is the dominant calcium binding protein in the cytoplasm (212,219), where it appears to play the major role in calcium transport from the terminal web to the basolateral membrane (190). The increase in calbindin levels in the cytosol following 1,25(OH)2D administration is blocked by protein synthesis inhibitors (210). Indeed, calbindin was the first protein discovered to be induced by vitamin D (219). Glenney and Glenney (212) observed that calbindin has a higher affinity for calcium than does calmodulin. The differential distribution of calmodulin and calbindin between microvillus and cytosol combined with the differences in affinity for calcium led Glenney and Glenney (212) to propose that in the course of calcium transport calcium flowed from calmodulin in the microvillus to calbindin in the cytosol with minimal change in the free calcium concentration in either location. However, the role of calbindin in intestinal calcium transport does not appear to be critical in that mice null for calbindin9k grow normally, have normal intestinal calcium transport, and their serum calcium levels and bone mineral content are equivalent to wildtype mice regardless of the calcium content of the diet (220). The CaATPase (PMCA1b) at the basolateral membrane and the sodium/calcium exchanger (NCX1) are responsible for removing calcium from the cell against the same steep electrochemical gradient as favored calcium entry at the brush border membrane (221). Related proteins are found in the renal distal tubule. As its name implies, the extrusion of calcium from the cell by the calcium pump requires ATP. This pump is a member of the PMCA family, and in the intestine the isoform PMCA1b is the major isoform found. This pump is induced by 1,25(OH)2D (222). Calmodulin activates the pump, but calbindin may do likewise (223). Deletion of Pmca1b reduces calcium absorption and blocks 1,25(OH)2D stimulation of such resulting in reduction in growth and bone mineralization (224)., Moreover, the deletion of protein 4.1R, which regulates PMCA1b expression in the intestine, results in decreased intestinal calcium transport (225). The role of NCX is not considered to be as important as PMCA1b for intestinal calcium transport (226).
The paracellular pathway has received less study, but accounts for the bulk of intestinal calcium transport in that the ileum accounts for around 80% of total calcium absorption essentially all by the paracellular pathway. Paracellular calcium absorption depends to a considerable extent on the gradient between the luminal calcium concentrations and the interstitial calcium concentrations. Thus, it is faster in the duodenum and upper jejunum than the ileum, but because the transit time in the ileum is so much longer than that of the upper GI tract, the ileum is where most of the calcium absorption takes place. Solvent drag plays a large part in moving calcium across the tight junctions between the epithelial cells (227) . Solvent flow follows the osmotic gradient which is maintained distal to the tight junction by the Na/K ATPase and sodium glucose cotransporter of the basolateral membrane which may be stimulated by 1,25(OH)2D (226,227). The tight junction itself provides both charge and size selectivity. The actomyosin ring around the tight junction contributes to the size selectivity (228). The claudins and occludins contribute to charge selectivity. Claudin 2, 12, 15 are negatively charged proteins enabling cations such as sodium and calcium to pass (229,230). 1,25(OH)2D stimulates the expression of claudins 2 and 12 (231). Prolactin stimulates claudin 15 expression, thought to contribute to the increased calcium absorption during pregnancy (232).
Although less studied, intestinal phosphate transport is also under the control of vitamin D. This was first demonstrated by Harrison and Harrison (233) in 1961. Active phosphate transport is greatest in the jejunum, in contrast to active calcium transport that is greatest in the duodenum. Cycloheximide blocks 1,25(OH)2D stimulated phosphate transport (234), indicating that protein synthesis is involved. Phosphate transport at both the brush border and basolateral membranes requires sodium. A sodium-phosphate transporter in the small intestine (NaPi-IIb), homologous to the type IIa sodium phosphate transporter in kidney, has been cloned and sequenced (235). Expression of NaPi-IIb is increased by 1,25(OH)2D (236). Transport of phosphate through the cytosol from one membrane to the other is poorly understood. However, cytochalasin B, a disrupter of microfilaments, has been shown to disrupt this process (237) suggesting that as for calcium, intracellular phosphate transport occurs in vesicles.
Bone
Nutritional vitamin D deficiency, altered vitamin D responsiveness such as vitamin D receptor mutations (hereditary vitamin D resistant rickets), and deficient production of 1,25(OH)2D such as mutations in the CYP27B1 gene (pseudo vitamin D deficiency) all have rickets as their main phenotype. This would suggest that vitamin D, and in particular 1,25(OH)2D, is of critical importance to bone. Furthermore, VDR are found in bone cells (238,239), and vitamin D metabolites have been shown to regulate many processes in bone. However, the rickets resulting from vitamin D deficiency or VDR mutations (or knockouts) can be corrected by supplying adequate amounts of calcium and phosphate either by infusions or orally [214-217]. Moreover, deletion of VDR from bone cells does not result in rickets (240). This would suggest either that vitamin D metabolites do not directly impact bone, or that substantial redundancy has been built into the system. However, arguing for a physiologically non-redundant direct action of vitamin D on bone is the development of osteoporosis and decreased bone formation in these VDR or CYP27B1 null mice not corrected by the high calcium/phosphate diet (241). A further complicating factor in determining the role of vitamin D metabolites in bone is the multitude of effects these metabolites have on systemic calcium homeostatic mechanisms which themselves impact on bone. The lack of vitamin D results in hypocalcemia and hypophosphatemia that as implied above is sufficient to cause rickets. Moreover, part of the skeletal phenotype in vitamin D deficiency is also due to the hyperparathyroidism that develops in the vitamin D deficient state as PTH has its own actions on bone and cartilage. Furthermore, within bone the vitamin D metabolites can alter the expression and/or secretion of a large number of skeletally derived factors including insulin like growth factor-1 (IGF-I) (242), its receptor (243), and binding proteins (244,245), transforming growth factor β (TGFβ) (246), vascular endothelial growth factor (VEGF) (247), interleukin-6 (IL-6) (248), IL-4 (249), and endothelin receptors (250) all of which can exert effects on bone of their own as well as modulate the actions of the vitamin D metabolites on bone. Understanding the impact of vitamin D metabolites on bone is additionally complicated by species differences, differences in responsiveness of bone and cartilage cells according to their states of differentiation, and differences in responsiveness in terms of the vitamin D metabolite being examined. Thus, the study of vitamin D on bone has had a complex history, and uncertainty remains as to how critical the direct actions of the vitamin D metabolites on bone are for bone formation and resorption.
Bone develops intramembranously (e.g., skull) or from cartilage (endochondral bone formation, e.g., long bones with growth plates). Intramembranous bone formation occurs when osteoprogenitor cells proliferate and produce osteoid, a type I collagen rich matrix. The osteoprogenitor cells differentiate into osteoblasts which then deposit calcium phosphate crystals into the matrix to produce woven bone. This bone is remodeled into mature lamellar bone. Endochondral bone formation is initiated by the differentiation of mesenchymal stem cells into chondroblasts that produce the proteoglycan rich type II collagen matrix. These cells continue to differentiate into hypertrophic chondrocytes that shift from making type II collagen to producing type X collagen. These cells also initiate the degradation and calcification of the matrix by secreting matrix vesicles filled with degradative enzymes such as metalloproteinases and phospholipases, alkaline phosphatase (thought to be critical for the mineralization process), and calcium phosphate crystals. Vascular invasion and osteoclastic resorption are stimulated by the production of VEGF and other chemotactic factors from the degraded matrix. The hypertrophic chondrocytes also begin to produce markers of osteoblasts such as osteocalcin, osteopontin, and type I collagen resulting in the initial deposition of osteoid. Moreover, at least some of these chondrocytes further differentiate (or trans differentiate) into osteoblasts (251). Terminal differentiation of the hypertrophic chondrocytes and the subsequent calcification of the matrix are markedly impaired in vitamin D deficiency leading to the flaring of the ends of the long bones and the rachitic rosary along the costochondral junctions of the ribs, classic features of rickets. Although supply of adequate amounts of calcium and phosphate may correct most of these defects in terminal differentiation and calcification, the vitamin D metabolites, 1,25(OH)2D and 24,25(OH)2D, have been shown to exert distinct roles in the process of endochondral bone formation.
The VDR makes its first appearance in the fetal rat at day 13 of gestation in the condensing mesenchyme of the vertebral column then subsequently in osteoblasts and the proliferating and hypertrophic chondrocytes by day 17 (252). However, fetal development is quite normal in vitamin D deficient rats (253) and VDR knockout mice (126) suggesting that vitamin D and the VDR are not critical for skeletal formation. Rickets develops postnatally, becoming most manifest after weaning. The impairment of endochondral bone formation observed in vitamin D deficiency is associated with decreased alkaline phosphatase activity of the hypertrophic chondrocytes (254), alterations in the lipid composition of the matrix (255) perhaps secondary to reduced phospholipase activity (256), and altered proteoglycan degradation (257) due to changes in metalloproteinase activity (257,258). Both 1,25(OH)2D and 24,25(OH)2D appear to be required for optimal endochondral bone formation (259). However, in the CYP24A1 knockout mouse, that fails to produce any 24-hydroxylated metabolites of vitamin D, the skeletal lesion is defective mineralization of intramembranous (not endochondral) bone. Furthermore, the skeletal abnormality appears to be due to high circulating 1,25(OH)2D levels in that crossing this mouse with one lacking the VDR corrects the problem (85). Whether this reflects species differences between mice and other species (most studies demonstrating the role of 24,25(OH)2D in bone and cartilage have used rats and chicks) remains unknown. Chondrocytes from the resting zone of the growth plate of rats tend to be more responsive to 24,25(OH)2D than 1,25(OH)2D, whereas the reverse is true for chondrocytes from the growth zone with respect to stimulation of alkaline phosphatase activity (260), regulation of phospholipase A2 (stimulation by 1,25(OH)2D, inhibition by 24,25(OH)2D) (261), changes in membrane fluidity (increased by 1,25(OH)2D, decreased by 24,25(OH)2D) (262), and stimulation of protein kinase C activity (263). These actions of 1,25(OH)2D and 24,25(OH)2D do not require the VDR and are non-genomic in that they take place with isolated matrix vesicles and membrane preparations from these cells (260). As discussed earlier membrane receptors for these vitamin D metabolites have been found in chondrocytes that may mediate these non-genomic actions (264). Osteoblasts also differ in their response to 1,25(OH)2D depending on their degree of maturation (265). In the latter stages of differentiation, rat osteoblasts respond to 1,25(OH)2D with an increase in osteocalcin production (266), but do not respond to 1,25(OH)2D in the early stages. Mice, however, differ from rats in that 1,25(OH)2D inhibits osteocalcin expression (266). Similarly, the effects of 1,25(OH)2D on alkaline phosphatase (267) and type I collagen (268) are inhibitory in the early stages of osteoblast differentiation but stimulatory in the latter stages (265). Osteopontin is better stimulated by 1,25(OH)2D in the early stages than the late stages of differentiation (265,269). Osteocalcin and osteopontin in human and rat cells have well described VDREs in their promoters (270-272) (the mouse does not) (273). However, alkaline phosphatase and the COL1A1 and COL1A2 genes producing type I collagen do not have clearly defined VDREs, so it remains unclear how these genes are regulated by 1,25(OH)2D. These maturation dependent effects of 1,25(OH)2D on bone cell function may explain the surprising ability of excess 1,25(OH)2D to block mineralization leading to hyperosteoidosis (274,275) as such doses may prevent the normal maturation of osteoblasts.
In addition to its role in promoting bone formation, 1,25(OH)2D also promotes bone resorption by increasing the number and activity of osteoclasts (276). Whether mature osteoclasts contain the VDR and are regulated directly by 1,25(OH)2D remains controversial (277,278), but the VDR in osteoclast precursors is not required for osteoclastogenesis. Rather, the stimulation of osteoclastogenesis by 1,25(OH)2D is mediated by osteoblasts. Rodan and Martin (279) originally proposed the hypothesis that osteoblasts were required for osteoclastogenesis, and the mechanism has now been elucidated (280). Osteoblasts produce a membrane associated protein known as RANKL (receptor activator of nuclear factor (NF)-kB ligand) that activates RANK on osteoclasts and their hematopoietic precursors. This cell-to-cell contact in combination with m-CSF also produced by osteoblasts stimulates the differentiation of precursors to osteoclasts, and promotes their activity. 1,25(OH)2D regulates this process by inducing RANKL (281) as does PTH, PGE2, and IL-11, all of which stimulate osteoclastogenesis. 1,25(OH)2D requires the VDR in osteoblasts for this purpose, although the other hormones and cytokines do not. Osteoblasts from Vdr knockout mice fail to support 1,25(OH)2D induced osteoclastogenesis, whereas osteoclast precursors from Vdr knockout mice can be induced by 1,25(OH)2D to form osteoclasts in the presence of osteoblasts from wildtype animals (282).
Kidney
The regulation of calcium and phosphate transport by vitamin D metabolites in the kidney has received less study than that in the intestine, but the two tissues have similar although not identical mechanisms. Eight grams of calcium are filtered by the glomerulus each day, and 98% of that is reabsorbed. Most is reabsorbed in the proximal tubule. This is a paracellular, sodium dependent process with little or no regulation by PTH and 1,25(OH)2D. Approximately 20% of calcium is reabsorbed in the thick ascending limb of the loop of Henle, 10-15% in the distal tubule, and 5% in the collecting duct (283). Regulation by vitamin D takes place in the distal tubule where calcium moves against an electrochemical gradient (presumably transcellular) in a sodium independent fashion (284). Phosphate, on the other hand, is approximately 80% reabsorbed in the proximal tubule, and this process is regulated by PTH (285). In parathyroidectomized (PTX) animals Puschett et al. (286-288)) demonstrated acute effects of 25OHD and 1,25(OH)2D on calcium and phosphate reabsorption. Subsequent studies indicated that PTH could enhance or was required for the stimulation of calcium and phosphate reabsorption by vitamin D metabolites (289,290).
The molecules critical for calcium reabsorption in the distal tubule appear to be the VDR, calbindin, TRPV5, and the BLM calcium pump (PMCA1b as in the intestine), a situation similar to the mechanism for calcium transport in the intestine. However, the calbindin in the kidney in most species is 28kDa, whereas the 9kDa form is found in the intestine in most species. The kidney has mostly TRPV5, whereas the intestine is primarily TRPV6. The calcium pump is the same isoform in both tissues (PMCA1b) although other forms of PMCA are also present. Calmodulin and a brush border myosin I like protein are also found in the kidney brush border, but their role in renal calcium transport has not been explored. VDR, calbindin, TRPV5, and PMCA1b colocalize in the distal tubule, but not all distal tubules contain this collection of proteins (201,202,291,292) suggesting that not all distal tubules are involved in calcium transport. 1,25(OH)2D upregulates the VDR (234), an action opposed by PTH (237). Calbindin is also induced by 1,25(OH)2D in the kidney(293,294). The activity of the calcium pump is increased by 1,25(OH)2D (295), but it is not clear that the protein itself is induced. The increased activity may be due to the induction of calbindin that increases its activity. The effect of 1,25(OH)2D on TRPV5 expression is stimulatory (205).
Phosphate reabsorption in the proximal tubule is mediated at the brush border by sodium dependent phosphate transporters (NaPi-2a and NaPi-2c) that rely on the baso-lateral membrane Na,K ATPase to maintain the sodium gradient that drives the transport process (296). It is not clear whether 1,25(OH)2D regulates the expression or activities of these transporters as it does in the intestine, although PTH clearly does. Like PTH, FGF23 blocks phosphate reabsorption, presumably by blocking NaPi-2a activity. Unlike PTH, FGF23 also blocks the renal production of 1,25(OH)2D, as discussed earlier. The link between phosphate reabsorption and 1,25(OH)2D production remains unclear.
TARGET TISSUE RESPONSES: NON-CALCIUM TRANSPORTING TISSUES
In addition to the its effects on tissues directly responsible for calcium homeostasis, 1,25(OH)2D regulates the function of a wide number of other tissues. These all contain the VDR. Regulation of differentiation and proliferation is one common theme; regulation of hormone secretion is another; regulation of immune function is the third. In most cases 1,25(OH)2D acts in conjunction with calcium. Selected examples follow.
Regulation of Hormone Secretion
PARATHYROID GLAND (PTH SECRETION)
As previously mentioned, PTH stimulates the production of 1,25(OH)2D. In turn 1,25(OH)2D inhibits the production of PTH (297,298). The regulation occurs at the transcriptional level. Within the promoter of the PTH gene is a region that binds the VDR and mediates the suppression of the PTH promoter by 1,25(OH)2D (162,293,299-303). However, there is substantial controversy about whether this site is a single half site (299) or a more classic DR3 (292), whether one VDRE is involved or two (300), whether only VDR binds (299,303), whether VDR/RXR heterodimers bind (162,300), or whether VDR partners with a different protein (301). Some of the differences may reflect different species, but the nature of PTH gene suppression by 1,25(OH)2D remains incompletely understood. Calcium alters the ability of 1,25(OH)2D to regulate PTH gene expression. Calcium is a potent inhibitor of PTH production and secretion, acting through the calcium sensing receptor (CaSR) on the plasma membrane of the parathyroid cell. 1,25(OH)2D induces the CaSR in the parathyroid gland making it more sensitive to calcium (304). Animals placed on a low calcium diet have an increase in PTH and 1,25(OH)2D levels indicating that the low calcium overrides the inhibition by 1,25(OH)2D on PTH secretion (305,306). One possible explanation involves the protein calreticulin that binds to nuclear hormone receptors including VDR at KXGFFKR sequences, and inhibits their activity (307,308). Low dietary calcium has been shown to increase calreticulin levels in the parathyroid gland (309). The ability of 1,25(OH)2D to inhibit PTH production and secretion has been exploited clinically in that 1,25(OH)2D and several of its analogs are used to prevent and/or treat secondary hyperparathyroidism associated with renal failure. The parathyroid gland also expresses CYP27B1 and so can produce its own 1,25(OH)2D that may act in an autocrine or paracrine fashion to regulate PTH production (310). As noted earlier, the parathyroid gland is one of several tissues expressing the megalin/cubilin complex potentially enabling it to take up 25OHD and other D metabolites still bound to DBP.
PANCREATIC BETA CELLS (INSULIN SECRETION)
1,25(OH)2D stimulates insulin secretion, although the mechanism is not well defined (311,312). VDR, CYP27B1 and calbindin-D28k are found in pancreatic beta cells (313-315), and studies using calbindin-D28k null mice have suggested that calbindin-D28k, by regulating intracellular calcium, can modulate depolarization-stimulated insulin release (316). Furthermore, calbindin-D28k, by buffering calcium, can protect against cytokine mediated destruction of beta cells (317). A number of mostly case control and observational studies have suggested that vitamin D deficiency contributes to increased risk for type 2 diabetes mellitus (318). Moreover, several randomized clinical trials evaluating the ability of vitamin D supplementation to prevent the progression of prediabetes to diabetes indicate that vitamin D has a modest protective effect especially in vitamin D deficient subjects (319,320).
FIBROBLAST GROWTH FACTOR (FGF23)
FGF23 is produced primarily by bone, and in particular by osteoblasts and osteocytes. 1,25(OH)2D3 stimulates this process, but the mechanism is not clear (322). Inasmuch as FGF23 inhibits 1,25(OH)2D production by the kidney, this feedback loop like that for PTH secretion maintains a balance in the levels of these important hormones. Mutations in the Phosphate regulating gene with Homologies to Endopeptidases on the X chromosome (PHEX) or FGF23 itself (which prevent its proteolysis) or conditions such as McCune-Albright disease and tumor induced osteomalacia in which FGF23 is overexpressed in the involved tissue led to hypophosphatemia and inappropriately low 1,25(OH)2D accompanied by osteomalacia. The role of PHEX, which was originally thought to cleave FGF23, in regulating FGF23 levels is not clear. In contrast mutations in UDP-N-acetyl-α-D galactosamine:polypeptide N-acetylgalactosaminyltransferase (GALNT3), which glycosylates FGF23, or in FGF23 which blocks this glycosylation result in inhibited FGF23 secretion leading to hyperphosphatemia, increased 1,25(OH)2D, and tumoral calcinosis (323).
Regulation of Proliferation and Differentiation
CANCER
1,25(OH)2D has been evaluated for its potential anticancer activity in animal and cell studies for nearly 40 years (324). The list of malignant cells that express VDR is now quite extensive, and the list of those same cells that express CYP27B1 is growing. The accepted basis for the promise of 1,25(OH)2D in the prevention and treatment of malignancy includes its antiproliferative, pro-differentiating effects on most cell types. The list of mechanisms proposed for these actions is extensive, and to some extent cell specific (325). Among these mechanisms 1,25(OH)2D has been shown to stimulate the expression of cell cycle inhibitors p21 and p27 (326) and the expression of the cell adhesion molecule E-cadherin (150), while inhibiting the transcriptional activity of β-catenin (150,327,328). In keratinocytes, 1,25(OH)2D has been shown to promote the repair of DNA damage induced by ultraviolet radiation (UVR) (329) (330), reduce apoptosis while increasing survival after UVR (331), and increase p53 (332). Epidemiologic evidence supporting the importance of adequate vitamin D nutrition (including sunlight exposure) for the prevention of a number of cancers (333-337) is extensive. Although numerous types of cancers show reduction (338), most attention has been paid to cancers of the breast, colon, and prostate. I (339) recently reviewed a number of meta-analyses of epidemiologic studies evaluating the association of vitamin D intake and/or 25OHD levels and the risk of developing these cancers. The data supporting a reduction in risk for developing colorectal cancer and breast cancer in premenopausal females with higher vitamin D intake or higher serum 25OHD levels were considerably stronger than that for the prevention of prostate cancer. Prospective randomized controlled trial data are limited. In a prospective 4 yr. trial with 1100iu vitamin D and 1400-1500 mg calcium originally designed to look at osteoporosis the authors showed a 77% reduction in cancers after excluding the initial year of study (340), including a reduction in both breast and colon cancers. In this study, vitamin D supplementation raised the 25OHD levels from a mean of 28.8ng/ml to 38.4ng/ml with no changes in the placebo or calcium only arms of the study. However, this was a relatively small study in which cancer prevention was not the primary outcome variable. A substantially larger trial involving over 25,000 subjects treated in a two by two design with vitamin D and/or omega 3 fatty acid did not find a benefit of vitamin supplementation with respect to cancer incidence but appears to have shown a beneficial effect on mortality (341). Trials of 1,25(OH)2D and its analogs for the treatment of cancer have been disappointing. In a small study involving 7 subjects with prostate cancer treated with doses of 1,25(OH)2D up to 2.5µg for 6-15 months, 6/7 showed a decrease in the rise of prostate specific antigen (PSA), a marker of tumor progression (342), and one patient showed a decline. However, hypercalciuria was common and limiting. A preliminary report of a larger study involving 250 patients with prostate cancer using 45µg 1,25(OH)2D weekly in combination with docetaxel demonstrated a non-significant decline in PSA, although survival was significantly improved (HR 0.67) (343). A larger follow-up study did not show increased survival (344). The incidence of either hypercalcemia or hypercalciuria was not reported. Most likely until an analog of 1,25(OH)2D is developed which is both efficacious and truly non hypercalcemic, treatment of cancer with vitamin D metabolites will remain problematic.
SKIN
Epidermal keratinocytes are the only cells in the body with the entire vitamin D metabolic pathway. As described earlier, production of vitamin D3 from 7-dehydrocholesterol takes place in the epidermis. However, the epidermis also contains CYP27A1 (345), the mitochondrial enzyme that 25-hydroxylates vitamin D, and CYP27B1 (40,47), the enzyme that produces 1,25(OH)2D from 25OHD. The CYP27B1 in keratinocytes is differently regulated than CYP27B1 in renal cells. Although PTH stimulates CYP27B1 activity in the keratinocyte, the mechanism appears to be independent of cAMP (346). Cytokines such as tumor necrosis factor-α and interferon-γ stimulate CYP27B1 activity (347,348). 1,25(OH)2D does not exert a direct effect on CYP27B1 expression in keratinocytes, but regulates 1,25(OH)2D levels by inducing CYP24A1 thus initiating the catabolism of 1,25(OH)2D (79). CYP27B1 is expressed primarily in the basal cells of the epidermis (50); as the cells differentiate the mRNA and protein levels of CYP27B1and its activity decline (349).
1,25(OH)2D regulates keratinocyte differentiation in part by modulating the ability of calcium to do likewise (350). Therefore, it is important to understand the actions of calcium on this cell prior to examining the influence of 1,25(OH)2D (351-356)(357). If keratinocytes are grown at calcium concentrations below 0.07mM, they continue to proliferate but either fail or are slow to develop intercellular contacts, stratify little if at all, and fail or are slow to form cornified envelopes. Acutely increasing the extracellular calcium concentration (Cao) above 0.1mM (calcium switch) leads to the rapid redistribution of desmoplakin, cadherins, integrins, catenins, plakoglobulin, vinculin, and actinin from the cytosol to the membrane where they participate in the formation of intercellular contacts. Calcium also stimulates the redistribution to the membrane of protein kinase Cα (PKCα) (358,359) and the tyrosine-phosphorylated p62 associated protein of ras GAP (360,361) where they further the calcium signaling process. These early events are accompanied by a rearrangement of actin filaments from a perinuclear to a radial pattern which if disrupted blocks the redistribution of these proteins and blocks the differentiation process. Within hours of the calcium switch keratinocytes switch from making the basal keratins K5 and K14 and begin making keratins K1 and K10 (356) followed, subsequently, by increased levels of profilaggrin (the precursor of filaggrin, an intermediate filament associated protein), involucrin and loricrin (precursors for the cornified envelope) (362,363). Loricrin, involucrin and other proteins (364) are cross linked into the insoluble cornified envelope by the calcium sensitive, membrane bound form of transglutaminase (365,366), which like involucrin and loricrin increases within 24 hours after the calcium switch (367). Within 1-2 days of the calcium switch cornified envelope formation is apparent (355,368), paralleling transglutaminase activation (369). The induction of these proteins represents a genomic action (likely indirect) of calcium as indicated by a calcium induced increase in mRNA levels and transcription rates (356,363,369,370). The relevance of calcium induced differentiation in vitro to the in vivo situation is indicated by the steep gradient of calcium within the epidermis, with the highest levels in the uppermost (most differentiated) nucleated layers (371). Current evidence for the importance of calcium in epidermal function is that barrier disruption, which results in increased proliferation, is associated with loss of the calcium gradient, whereas increasing the calcium concentration in the epidermis with sonophoresis stimulates lamellar body secretion (372-376).
The keratinocyte senses calcium via a seven transmembrane domain, G protein coupled receptor (CaSR) (377) originally cloned from the parathyroid cell by Brown et al (378,379). Knocking out the CaSR blocks calcium induced differentiation in vitro (380,381) and in vivo (382). However, keratinocytes also produce an alternatively spliced variant of the CaSR as they differentiate (383). This variant CaSR lacks exon 5 and so would be missing residues 461-537 in the extracellular domain. A mouse model in which the full length CaSR has been knocked out continues to produce the alternatively spliced form of CaSR, but its epidermis contains lower levels of the terminal differentiation markers loricrin and profilaggrin, and keratinocytes from these mice fail to respond normally to calcium (383) consistent with the results when the full length calcium receptor was deleted in vitro (380,381). We have produced a conditional knockout of the CaSR allowing us to delete CaSR in the tissue of choice using cell specific cre recombinases that avoids the problem with the original global knockout (384). When the CaSR is deleted specifically in the keratinocyte, this mouse has a reduction in epidermal differentiation and barrier repair (382), but unlike the global knockout does not have abnormalities in overall calcium homeostasis, and rather than showing an increased calcium gradient in the epidermis has a blunted one. The conditional knockout mouse also lacks the alternatively spliced CaSR.
Inositol 1,4,5 tris phosphate (IP3) and diacylglycerol levels increase within seconds to minutes after the calcium switch implicating activation of the phospholipase C (PLC) pathway (385,386). Similar to intracellular calcium levels (Cai), the levels of inositol phosphates (IPs) remain elevated for hours after the calcium switch. The prolonged increase in IPs after the calcium switch may contribute to the plateau phase of Cai elevation and a prolonged elevation of diacylglycerol (DG) that would stimulate the protein kinase C (PKC) pathway. This prolonged increase in IPs appears to be due to calcium induction and activation of PLC (154,386,387), especially PLC-γ1. Activation of PLC-γ1 by calcium involves a chain of events involving src kinase activation of phosphatidyl inositol 3 kinase and phosphatidyl inositol 4 phosphate 5 kinase 1α within the context of a membrane complex with E-cadherin leading to the formation of phosphatidyl inositol tris phosphate in the membrane which activates PLC-γ1 via its PH domain (388). Phosphorylation of PLC-γ1 is not part of its activation by calcium unlike its activation by EGF (389). Knocking out Plcg1 blocks the ability of calcium to increase Cai and to induce involucrin and transglutaminase (387). Thus, like CaSR, PLC-γ1 is critical for the ability of calcium to regulate keratinocyte differentiation.
Phorbol esters, which bind to and activate PKC, are well known tumor promoters in skin However, the initial effects of phorbol esters in vitro are to promote differentiation in cells grown in low calcium (358,390,391), effects which are potentiated by calcium (383). Phorbol esters stimulate PKC, and PKC inhibitors block the ability of both calcium and phorbol esters to promote differentiation (391). Phorbol esters as well as calcium stimulate the expression of both keratin 1 and involucrin gene constructs each of which contains an AP-1 site within the calcium response element (CaRE) of the promoter for these genes (392,393). If the AP-1 site within the CaRE is mutated, neither calcium nor phorbol esters are effective (392,393). These CaREs also contain VDREs (DR3), which at least in the involucrin gene has been shown to mediate 1,25(OH)2D regulation of this gene (394). Phorbol esters do not reproduce all the actions of calcium on the keratinocyte, and vice versa, but cross talk between their signaling pathways is clearly present.
The observation that 1,25(OH)2D induces keratinocyte differentiation was first made by Hosomi et al. (395) and provided a rationale for the previous and unexpected finding of 1,25(OH)2D receptors in the epidermis (396). 1,25(OH)2D increases the mRNA and protein levels for involucrin and transglutaminase, and promotes CE formation at subnanomolar concentrations in preconfluent keratinocytes (370,397-399). Calcium affects the ability of 1,25(OH)2D to stimulate keratinocyte differentiation, and vice versa. Calcium in the absence of 1,25(OH)2D and 1,25(OH)2D at low (0.03mM) calcium raise the mRNA levels for involucrin and transglutaminase in a dose dependent fashion by stimulating gene expression. The stimulation of mRNA levels by calcium and 1,25(OH)2D is synergistic at early time points; however, longer periods of incubation lead to a paradoxical fall in the mRNA levels for these proteins. This is due to the fact that although transcription is increased by calcium and 1,25(OH)2D, stability of the mRNA is reduced in cells incubated with calcium and 1,25(OH)2D.
The transcriptional regulation by 1,25(OH)2D is both direct and indirect. Several genes contain VDREs (e.g. involucrin), but VDREs have not been found in all genes that are regulated by 1,25(OH)2D. Inhibition of PKC activity or mutation of the AP-1 site in the CaRE of the involucrin gene also blocks the ability of 1,25(OH)2D to regulate expression of involucrin (394). The ability of 1,25(OH)2D to increase intracellular calcium (Cai) (298) accounts for at least part of the ability of 1,25(OH)2D to induce differentiation. A rapid (presumably nongenomic) effect of 1,25(OH)2D on Cai has been described (400), although this response is controversial (398). Our studies indicate that the ability of 1,25(OH)2D to increase Cai requires time and gene transcription. 1,25(OH)2D increases CaSR mRNA levels and prevents their fall in cells grown in 0.03mM calcium (401). This results in an enhanced Cai response to extracellular calcium (Cao). 1,25(OH)2D also induces the family of PLCs (402). PLC-γ1 contains a VDRE in its promoter (154), which unlike the usual VDRE is a DR6 which binds VDR/RAR rather than VDR/RXR. Knocking out PLCG1 blocks 1,25(OH)2D induced differentiation (403) as well as calcium induced differentiation mentioned earlier. The other PLCs have not been studied as extensively, but are likely to show similar means of regulation by 1,25(OH)2D.
Our current working model for the mechanisms by which calcium and 1,25(OH)2D regulate keratinocyte differentiation is shown in figure 8. The keratinocyte expresses a CaSR that by coupling to and activating PLC controls the production of two important second messengers, IP3 and DG. PLC-β is likely to be activated acutely by CaSR via a G protein coupled mechanism, whereas PLC-γ1 is activated acutely by calcium stimulated non receptor tyrosine kinases and subsequently by PIP3 in the membrane. Both PLCs are induced by calcium and 1,25(OH)2D. IP3 stimulates the release of calcium from intracellular stores thus raising Cai. The initial release of calcium from these stores activates the Stim1/Orai1 channel in the membrane (404) that may stimulate proliferation of the basal keratinocytes and initiate their movement out of the basal layer. The increase in Cai and DG stimulates the activation of critical PKCs and their translocation to membrane receptors (RACK). PKC-α appears to be the most critical PKC for the subsequent events triggered by calcium in the keratinocyte, although PKCδ has also been implicated. Activated PKC leads to the induction and activation of AP-1 transcription factors which regulate the transcription of a number of genes including keratin 1, transglutaminase, involucrin, loricrin, and profilaggrin required for the differentiation process. Activation of the CaSR also activates the RhoA kinase leading to activation of src kinases which by phosphorylating various catenins leads to the formation of the Ecadherin/catenin complex in the membrane (405). This complex recruits both PI3K and PIP5K1α required to maintain the PIP2 and PIP3 levels in the membrane (357). PIP3 activates PLC-γ, that is in turn activates the TRPC channels in the membrane to enable the prolonged increase in Cai required for differentiation (406). 1,25(OH)2D, which is produced by the keratinocyte in a highly regulated fashion, modulates calcium regulated differentiation at several steps. First, 1,25(OH)2D increases CaSR expression, thus making the cell more responsive to calcium. Secondly, 1,25(OH)2D induces all the PLCs again increasing the responsiveness of the cell to calcium. Finally, 1,25(OH)2D has a direct effect on the transcription of the genes such as involucrin. The net result is that both calcium and 1,25(OH)2D promote keratinocyte differentiation through interactive mechanisms.
Figure 8.
A model of 1,25(OH)2D and calcium regulated keratinocyte differentiation. The G-protein coupled calcium receptor (CaSR) when activated by extracellular calcium activates Gα as described in the legend to figure 6. Gα stimulates PLC mediated hydrolysis of PIP2 to IP3 and DG. IP3 releases Cai from intracellular stores, and DG activates PKC. Depletion of intracellular calcium stores leads to influx of calcium across store operated calcium channels. PKC stimulation leads to activation of AP-1 transcription factors which along with calcium and 1,25(OH)2D activated transcription factors stimulate the expression of genes essential for the differentiation process. 1,25(OH)2D regulates this process by inducing CaSR and PLC as well as genes essential for cornified envelope formation such as involucrin and transglutaminase.
The VDR is also critical for hair follicle (HF) cycling. Unlike epidermal differentiation, hair follicle cycling is not dependent on 1,25(OH)2D. Alopecia is a well described characteristic of mice and humans lacking VDR (125,126,407) due to failure to regenerate the cycling lower portion of the HF after the initial developmental cycle is completed. Deletion of CYP27B1 (408) and CaSR (382) do not result in alopecia. Cianferotti et al. (409) attributed the loss of HF cycling in VDR null mice to a gradual loss of the proliferative potential in the stem cells of the HF bulge region. However, this conclusion has been challenged by Palmer et al. (410), who attributed the failure of HF cycling in the VDR null mouse in part to a failure of the progeny of these stem cells to migrate out of the bulge rather than their loss of proliferative potential suggesting a loss of activation. The role of VDR in the stem cells that regulate both HF cycling and epidermal regeneration is also important in the skin wound healing process. When the skin is wounded the progeny of stem cells from all regions of the HF and interfollicular epidermis (IFE) contribute at least initially (411,412), although the stem cells in the IFE make the most lasting contribution. Tian et al. (413) observed that topical 1,25(OH)2D enhanced wound healing, suggesting that unlike HF cycling, the wound repair required this VDR ligand. Luderer et al. (414) observed that in the global VDRKO, there was a reduction in TGFβ signaling in the dermis, and subsequently demonstrated that the VDR in macrophages but not in keratinocytes was responsible for macrophage recruitment during the inflammatory phase of cutaneous wound healing (415). Our studies have focused on the VDR in epidermal keratinocytes. We have observed that re-epithelialization by the keratinocytes over the wound is impaired when the deletion of VDR from keratinocytes is accompanied by either a low calcium diet or a deletion of the CaSR (416). Thus like the role of calcium and CaSR in vitamin D regulated keratinocyte differentiation so a similar synergism is seen in wound healing. These results are consistent with the loss of E-cadherin/catenin complex formation in the VDRKO keratinocyte, a complex that maintains stem cells in their niches (417), regulates when stem cell division is symmetric (to maintain stem cell numbers) or asymmetric (initiating differentiation) (418), and is essential for the ability of keratinocytes to migrate as a sheet to re-epithelialize the wound (419). As noted previously calcium and the CaSR along with 1,25(OH)2D and VDR are required for E-cadherin/catenin complex formation during the differentiation process and so are involved in enabling its role in wound healing (420).
Immune System
The potential role for vitamin D and its active metabolite 1,25(OH)2D3 in modulating the immune response has long been recognized since the discovery of vitamin D receptors (VDR) in macrophages, dendritic cells (DC), and activated T and B lymphocytes, the ability of macrophages and DC as well as activated T and B cells to express CYP27B1, and the ability of 1,25(OH)2D3 to regulate the proliferation and function of these cells. While these are the key cells mediating the adaptive immune response, 1,25(OH)2D, VDR, and CYP27B1 are also expressed in a large number of epithelial cells which along with the aforementioned members of the adaptive immune response contribute to host defense by their innate immune response. The totality of the immune response involves both types of responses in complex interactions involving numerous cytokines. The regulation of these different responses and their interactions by 1,25(OH)2D3 is nuanced. In general, 1,25(OH)2D3 enhances the innate immune response primarily via its ability to stimulate cathelicidin, an antimicrobial peptide important in defense against invading organisms, whereas it inhibits the adaptive immune response primarily by inhibiting the maturation of dendritic cells (DC) important for antigen presentation, reducing T cell proliferation, and shifting the balance of T cell differentiation from the Th1 and Th17 pathways to Th2 and Treg pathways. Inflammatory autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis involve Th17 activation, a cell that expresses RANKL, and so can drive osteoclastogenesis leading to bone loss.
ADAPTIVE IMMUNE RESPONSE
The adaptive immune response is initiated by cells specialized in antigen presentation, DC and macrophages in particular, activating the cells responsible for subsequent antigen recognition, T and B lymphocytes. These cells are capable of a wide repertoire of responses that ultimately determine the nature and duration of the immune response. Activation of T and B cells occurs after a priming period in tissues of the body, e.g., lymph nodes, distant from the site of the initial exposure to the antigenic substance, and is marked by proliferation of the activated T and B cells accompanied by post translational modifications of immunoglobulin production that enable the cellular response to adapt specifically to the antigen presented. Importantly, the type of T cell activated, CD4 or CD8, or within the helper T cell class Th1, Th2, Th17, Treg, and subtle variations of those, is dependent on the context of the antigen presented by which cell and in what environment. Systemic factors such as vitamin D influence this process. Vitamin D in general exerts an inhibitory action on the adaptive immune system. 1,25(OH)2D3 decreases the maturation of DC as marked by inhibited expression of the costimulatory molecules HLA-DR, CD40, CD80, and CD86, decreasing their ability to present antigen and so activate T cells (421). Furthermore, by suppressing IL-12 production, important for Th1 development, and IL-23 and IL-6 production important for Th17 development and function, 1,25(OH)2D3 inhibits the development of Th1 cells capable of producing IFN-γ and IL-2, and Th17 cells producing IL-17 (422). These actions prevent further antigen presentation to and recruitment of T lymphocytes (role of IFN-γ), and T lymphocyte proliferation (role of IL-2). Suppression of IL-12 increases the development of Th2 cells leading to increased IL-4, IL-5, and IL-13 production, which further suppresses Th1 development shifting the balance to a Th2 cell phenotype. Treatment of DCs with 1,25(OH)2D3 can also induce CD4+/CD25+ regulatory T cells (Treg) cells (423) as shown by increased FoxP3 expression, critical for Treg development. These cells produce IL-10, which suppresses the development of the other Th subclasses. Treg are critical for the induction of immune tolerance (424). In addition, 1,25(OH)2D3 alters the homing of properties of T cells for example by inducing expression of CCR10, the receptor for CCL27, a keratinocyte specific cytokine, while suppressing that of CCR9, a gut homing receptor (425). The actions of 1,25(OH)2D3 on B cells have received less attention, but recent studies have demonstrated a reduction in proliferation, maturation to plasma cells and immunoglobulin production (426).
1,25(OH)2D3 has both direct and indirect effects on regulation of a number of cytokines involved with the immune response (review in (427)). TNF has a VDRE in its promoter to which the VDR/RXR complex binds. 1,25(OH)2D3 both blocks the activation of NFκB via an increase in IκBα expression and impedes its binding to its response elements in the genes such as IL-8 and IL-12 that it regulates. 1,25(OH)2D3 has also been shown to bring an inhibitor complex containing histone deacetylase 3 (HDAC3) to the promoter of rel B, one of the members of the NFκB family, thus suppressing gene expression. Thus, TNF/NFkB activity is markedly impaired by 1,25(OH)2D3 at multiple levels. In VDR null fibroblasts, NFκB activity is enhanced. Furthermore, 1,25(OH)2D3 suppresses IFNγ, and a negative VDRE has been found in the IFNγ promoter. GM-CSF is regulated by VDR monomers binding to a repressive complex in the promoter of this gene, competing with nuclear factor of T cells 1(NFAT1) for binding to the promoter.
The ability of 1,25(OH)2D3 to suppress the adaptive immune system appears to be beneficial for a number of conditions in which the immune system is directed at self—i.e. autoimmunity (review in (428)). In a number of experimental models including inflammatory arthritis, psoriasis, autoimmune diabetes (e.g., NOD mice), systemic lupus erythematosis (SLE), experimental allergic encephalitis (EAE) (a model for multiple sclerosis), inflammatory bowel disease (IBD), prostatitis, and thyroiditis VDR agonist administration has prevented and/or treated the disease process. As will be discussed later, a number of these conditions are associated with bone loss either directly (e.g., inflammatory arthritis) or indirectly presumably via increased serum levels of inflammatory cytokines. These actions of 1,25(OH)2D3 were originally ascribed to inhibition of Th1 function, but Th17 cells have also been shown to play important roles in a number of these conditions including psoriasis (321), experimental colitis (422), and rheumatoid arthritis (429), conditions that respond to 1,25(OH)2D3 and its analogs. Although few prospective, randomized, placebo-controlled trials in humans have been performed, epidemiologic and case control studies indicate that a number of these diseases in humans are favorably impacted by adequate vitamin D levels. For example, the incidence of multiple sclerosis correlates inversely with 25OHD levels and vitamin D intake, and early studies suggested benefit in the treatment of patients with rheumatoid arthritis and multiple sclerosis with VDR agonists (427,428). Similarly, IBD is associated with low vitamin D levels (430). Children who are vitamin D deficient have a higher risk of developing type 1 diabetes mellitus, and supplementation with vitamin D during early childhood reduces the risk of developing type 1 diabetes (review in (421)). In VDR null mice myelopoiesis and the composition of lymphoid organs are normal, although a number of abnormalities in the immune response have been found. Some of the abnormalities in macrophage function and T cell proliferation in response to anti-CD3 stimulation in these animals could be reversed by placing the animals on a high calcium diet to normalize serum calcium (431), indicating the important role of calcium in vitamin D regulated immune function as in skeletal development and maintenance, hormone regulation, and keratinocyte differentiation. Other studies have noted an increased number of mature DCs in the lymph nodes of VDR null mice, which would be expected to promote the adaptive immune response (432). Somewhat surprisingly, RANKL also increases the number and retention of DCs in lymph nodes (433) suggesting that at least this mechanism is not mediated via the RANKL/RANK system in VDR null mice, which I will discuss at length subsequently. In contrast to these inhibitory actions of 1,25(OH)2D3, Th2 function as indicated by increased IgE stimulated histamine from mast cells is increased in VDR null mice (434). The IL-10 null mouse model of IBD shows an accelerated disease profile when bred with the VDR null mouse with increased expression of Th1 cytokines (435). Surprisingly, despite a reduction in natural killer T cells and Treg cells and a decreased number of mature DCs, VDR null mice bred with NOD mice do not show accelerated development of diabetes (436). Part of the difference in tissue response in VDR null mice may relate to differences in the ability of 1,25(OH)2D3 to alter the homing of T cells to the different tissues (425). In allergic airway disease (asthma) Th2 cells, not Th1 cells, dominate the inflammatory response. 1,25(OH)2D3 administration to normal mice protected these mice from experimentally induced asthma in one study, blocking eosinophil infiltration, IL-4 production, and limiting histologic evidence of inflammation (437). However, a study with VDR null mice using a comparable method of inducing asthma showed that lack of VDR also protected the mice from an inflammatory response in their lungs (438). In an extension of this study the investigators showed that wildtype (WT) splenocytes were only minimally successful at restoring experimental airway inflammation to VDR null mice, whereas splenocytes from these mice were able to transfer experimental airway inflammation to the unprimed WT host (439). Thus, the impact of vitamin D signaling on adaptive immunity depends on the specifics of the immune response being evaluated.
Inhibition of the adaptive immune response may also have benefit in transplantation procedures (440). In experimental allograft models of the aorta, bone, bone marrow, heart, kidney, liver, pancreatic islets, skin, and small bowel VDR agonists have shown benefit generally in combination with other immunosuppressive agents such as cyclosporine, tacrolimus, sirolimus, and glucocorticoids (440). Much of the effect could be attributed to a reduction in infiltration of Th1 cells, macrophages and DC into the grafted tissue associated with a reduction in chemokines such as CXCL10, CXCL9, CCL2, and CCL5. CXCL10, the ligand for CXCR3, may be of particular importance for acute rejection in a number of tissues, whereas CXCL9 as well as CXCL10 (both CXCR3 ligands) may be more important for chronic rejection at least in the heart and kidney, respectively. Although there are no prospective trials of the use of VDR agonists in transplant patients, several retrospective studies in patients with renal transplants treated with 1,25(OH)2D3 have suggested benefit with respect to prolonged graft survival and reduced numbers of acute rejection episodes.
Suppression of the adaptive immune system may not be without a price. Several publications have demonstrated that for some infections including Leishmania major (441) and toxoplasmosis (442), 1,25(OH)2D3 promotes the infection (442), while the mouse null for VDR is protected (441). This may be due at least in part to loss of IFNγ stimulation of ROS and NO production required for macrophage antimicrobial activity (441). Furthermore, atopic dermatitis, a disease associated with increased Th2 activity (443), and allergic airway disease, likewise associated with increased Th2 activity, (437-439), may be aggravated by 1,25(OH)2D3 and less severe in animals null for VDR.
THE INNATE IMMUNE RESPONSE
The innate immune response involves the activation of toll-like receptors (TLRs) in polymorphonuclear cells (PMNs), monocytes and macrophages as well as in a number of epithelial cells including those of the epidermis, gingiva, intestine, vagina, bladder and lungs (review in (444)). There are 10 functional TLRs in human cells (of 11 known mammalian TLRs). TLRs are an extended family of host noncatalytic transmembrane pathogen-recognition receptors that interact with specific membrane patterns (PAMP) shed by infectious agents that trigger the innate immune response in the host. A number of these TLRs signal through adapter molecules such as myeloid differentiation factor-88 (MyD88) and the TIR-domain containing adapter inducing IFN-β (TRIF). MyD88 signaling includes translocation of NFkB to the nucleus, leading to the production and secretion of a number of inflammatory cytokines. TRIF signaling leads to the activation of interferon regulatory factor-3 (IRF-3) and the induction of type 1 interferons such as IFNβ. MyD88 mediates signaling from TLRs 2, 4, 5, 7 and 9, whereas TRIF mediates signaling from TLR 3 and 4. TLR1/2, TLR4, TLR5, TLR2/6 respond to bacterial ligands, whereas, TLR3, TLR7, and TLR 8 respond to viral ligands. The TLR response to fungi is less well defined. CD14 serves as a coreceptor for a number of these TLRs. Activation of TLRs leads to the induction of antimicrobial peptides (AMPs) and reactive oxygen species, which kill the organism. Among these AMPs is cathelicidin. Cathelicidin plays a number of roles in the innate immune response. The precursor protein, hCAP18, must be cleaved to its major peptide LL-37 to be active. In addition to its antimicrobial properties, LL-37 can stimulate the release of cytokines such as IL-6 and IL-10 through G protein coupled receptors, and IL-18 through ERK/P38 pathways, stimulate the EGF receptor leading to activation of STAT1 and 3, induce the chemotaxis of neutrophils, monocytes, macrophages, and T cells into the skin, and promote keratinocyte proliferation and migration (445). The expression of this antimicrobial peptide is induced by 1,25(OH)2D3 in both myeloid and epithelial cells (446,447). In addition, 1,25(OH)2D3 induces the coreceptor CD14 in keratinocytes(448). Stimulation of TLR2 by infectious organisms like tuberculosis in macrophages (449) or stimulation of TLR2 in keratinocytes by wounding the epidermis (448) results in increased expression of CYP27B1, which in the presence of adequate substrate (25OHD) stimulates the expression of cathelicidin. Lack of substrate (25OHD) or lack of CYP27B1 blunts the ability of these cells to respond to a challenge with respect to cathelicidin and/or CD14 production (447-449). In diseases such as atopic dermatitis, the production of cathelicidin and other antimicrobial peptides (AMPs) is reduced, predisposing these patients to microbial superinfections (450). Th2 cytokines such as IL-4 and 13 suppress the induction of AMPs(451). Since 1,25(OH)2D3 stimulates the differentiation of Th2 cells, in this disease 1,25(OH)2D3 administration may be harmful. An important role of these AMPs besides their antimicrobial properties is to help link the innate and adaptive immune response. This interplay is well demonstrated in SARS-CoV-19 infections in which a dysfunctional and/or delayed innate immune response can lead to an unchecked adaptive immune response resulting in a massive release of proinflammatory cytokines, the “cytokine storm”, leading to destruction of the lungs and death (452). Patients with vitamin D deficiency appear to be more vulnerable to this infection (453).
Although many cells are capable of the innate immune response including bone cells, most studies have focused on the macrophage and the keratinocyte. Vitamin D regulation of the innate immune response in these two cell types is comparable, but differences exist.
Macrophages
The importance of adequate vitamin D nutrition for resistance to infection has long been appreciated but poorly understood. This has been especially true for tuberculosis. Indeed, prior to the development of specific drugs for the treatment of tuberculosis, getting out of the city into fresh air and sunlight was the treatment of choice. In a recent survey of patients with tuberculosis in London (454) 56% had undetectable 25OHD levels, and an additional 20% had detectable levels but below 9 ng/ml (22 nM). In 1986 Rook et al. (455) demonstrated that 1,25(OH)2D3 could inhibit the growth of Mycobacterium tuberculosis. The mechanism for this remained unclear until the publication by Liu et al. (449) of their results in macrophages. They observed that activation of the Toll-like receptor TLR2/1 by a lipoprotein extracted from M. tuberculosis reduced the viability of intracellular M. tuberculosis in human monocytes and macrophages concomitant with increased expression of the VDR and of CYP27B1 in these cells. Killing of M. tuberculosis occurred only when the serum in which the cells were cultured contained adequate levels of 25OHD, the substrate for CYP27B1. This provided clear evidence for the importance of vitamin D nutrition (as manifested by adequate serum levels of 25OHD) in preventing and treating this disease, and demonstrated the critical role for endogenous production of 1,25(OH)2D3 by the macrophage to enable its antimycobacterial capacity. Activation of TLR2/1 or directly treating these cells with 1,25(OH)2D3 induced the antimicrobial peptide cathelicidin, which is toxic for M. tuberculosis. If induction of cathelicidin is blocked as with siRNA, the ability of 1,25(OH)2D3 to enhance the killing of M. tuberculosis is prevented (456). Furthermore, 1,25(OH)2D3 also induces the production of reactive oxygen species which if blocked likewise prevents the anti-mycobacterial activity of 1,25(OH)2D3 treated macrophages (457). The murine cathelicidin gene lacks a known VDR response element in its promoter, and so might not be expected to be induced by 1,25(OH)2D3 in mouse cells, yet 1,25(OH)2D3 stimulates antimycobacterial activity in murine macrophages. Murine macrophages, unlike human macrophages, utilize inducible nitric oxide synthase (iNOS) for their TLR and 1,25(OH)2D3 mediated killing of M. tuberculosis (457,458). Clinical trials attempting to treat tuberculosis patients with high levels of vitamin D have shown mixed results (459)(460).
Keratinocytes
Cathelicidiin and CD14 expression in epidermal keratinocytes is induced by 1,25(OH)2D3 (445,448). In these cells butyrate, which by itself has little effect, potentiates the ability of 1,25(OH)2D3 to induce cathelicidin (461). Keratinocytes treated with 1,25(OH)2D3 are substantially more effective in killing Staphyococcus aureus than are untreated keratinocytes. Wounding the epidermis induces the expression of TLR2 and that of its co-receptor CD14 and cathelicidin (448). This does not occur in mice lacking CYP27B1 (448). Unlike macrophages, 1,25(OH)2D3 stimulates TLR2 expression in keratinocytes as well as in the epidermis when applied topically (448) providing a feed forward loop to amplify the innate immune response. Wounding also increases the expression of CYP27B1. This may occur as a result of increased levels of cytokines such as TNF-α and IFN-γ, both of which we have shown stimulate 1,25(OH)2D3 production, as well as by TGF-β and the TLR2 ligand Malp-2 (448). When the levels of VDR or one of its principal coactivators, SRC3, are reduced using siRNA technology, the ability of 1,25(OH)2D3 to induce cathelicidin and CD14 expression in human keratinocytes is markedly blunted (461).
Other Tissues
The VDR is widespread (127,462) (reviews). In some of these tissues the functional significance of the VDR and/or the effect of 1,25(OH)2D are unclear. Since several of the functions regulated by 1,25(OH)2D in some of these tissues may have clinical relevance, this section will focus on a select number of these tissues.
HEART
A reduction in contractility has been observed in vitamin D deficient animals (463). This may be due to lack of vitamin D or the accompanying hypocalcemia and hypophosphatemia. However, in vitro 1,25(OH)2D stimulates calcium uptake by cardiac muscle cells (464,465). In addition, 1,25(OH)2D inhibits the expression of atrial naturetic factor, one of the few genes with a negative VDRE in its promoter (466). Deletion of the VDR specifically in cardiac muscle leads to hypertrophy and fibrosis (467). Low circulating levels of 25OHD are associated with increased risk of myocardial infarction in men [436]. However, a large randomized clinical trial failed to show a protective effective of vitamin D supplementation to individuals with normal levels of 25OHD with respect to cardiovascular disease (341)
SKELETAL MUSCLE
Proximal muscle weakness is a hallmark of vitamin D deficiency, and reduced high energy substrates (ATP, creatinine phosphate) have been observed in that condition (468). Myoblasts contain VDR, although the expression of VDR in mature muscle cells is controversial. Muscle weakness may reflect the lower levels of calcium and phosphate rather than a reduction in 1,25(OH)2D. However, evidence for a direct role of 1,25(OH)2D and VDR in muscle function is increasing (469). Moreover, 1,25(OH)2D may have actions on muscle that do not require the VDR, at least the genomic functions of VDR. The Boland laboratory (470) has demonstrated acute effects of 1,25(OH)2D on calcium uptake, PLC, PLA2, PLD, PKC, and adenylate cyclase activities, all of which may alter muscle function.
PITUITARY
VDR have been found primarily in thyrotropes in vivo and in GH and prolactin secreting cell lines in vitro (471,472). 1,25(OH)2D increases TRH stimulated TSH secretion by a mechanism involving increased Cai and IP3 production (473,474), suggesting that induction of PLC by 1,25(OH)2D may be involved.
BREAST
The breast contains VDR (475), and vitamin D plays a role in normal breast development (476). Moreover, breast cancer cells also contain VDR (477), and 1,25(OH)2D and its analogs reduce their proliferation in vivo and in vitro (478,479). This has obvious clinical implications for the treatment of breast cancer.
LIVER
Low levels of VDR have been found in the liver, particularly in stellate cells (480,481). Hepatic regeneration is impaired in vitamin D deficient animals, even when the serum calcium is normalized by a high calcium diet (482), suggesting a role for 1,25(OH)2D in hepatic cell growth and in the prevention of hepatic fibrosis (481).
LUNG
VDR have been found in type II epithelial pneumocytes (483). 1,25(OH)2D stimulates their maturation including increased phospholipid production and surfactant release [437].These results are consistent with the abnormal alveolar development observed in pups born to vitamin D deficient mothers (484). In addition 1,25(OH)2D stimulates the innate immune response in bronchial epithelial cells and may provide protection in patients with cystic fibrosis with recurrent lung infections as well as in patient with Covid-19 infections (452,485) as discussed previously.
REFERENCES
- 1.
- Whistler D. De morbo puerli anglorum, quem patrio ideiomate indigenae vocant "the rickets". Journal of History of Medicine. 1645;5:397–415.
- 2.
- Mellanby E. An experimental investigation on rickets. Lancet. 1919;1:407–412.
- 3.
- McCollum EV, Simmonds N, Becker JE, Shipley PG. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. Journal of Biological Chemistry. 1922;53:293–298. [PubMed: 11991957]
- 4.
- Steenbock H, Black A. Fat-soluble vitamins. XVII. The induction of growth-promoting and calcifying properties in a ration by exposure to ultraviolet light. Journal of Biological Chemistry. 1924;61:405–422.
- 5.
- Huldshinsky K. Heilung von rachitis durch kunstalich hohensonne. Deut Med Wochenschr. 1919;45:712–713.
- 6.
- Hess AF, Unger LF. Cure of infantile rickets by sunlight. Journal of The American Medical Association. 1921;77:39.
- 7.
- Askew FA, Bourdillon RB, Bruce HM, Jenkins RGC, Webster TA. The distillation of vitamin D. Proceedings of the Royal Society. 1931;B107:76–90.
- 8.
- Windaus A, Schenck F, von Werder F. Uber das antirachitisch wirksame bestrahlungs-produkt aus 7-dehydro-cholesterin. Hoppe-Seylers Z Physiological Chemistry. 1936;241:100–103.
- 9.
- Prabhu AV, Luu W, Sharpe LJ, Brown AJ. Cholesterol-mediated Degradation of 7-Dehydrocholesterol Reductase Switches the Balance from Cholesterol to Vitamin D Synthesis. The Journal of biological chemistry. 2016;291(16):8363–8373. [PMC free article: PMC4861412] [PubMed: 26887953]
- 10.
- Holick MF, McLaughlin JA, Clark MB, Doppelt SH. Factors that influence the cutaneous photosynthesis of previtamin D3. Science. 1981;211:590–593. [PubMed: 6256855]
- 11.
- Holick MF, McLaughlin JA, Clark MB, Holick SA. J.T. PJ, Anderson RR, Blank IH, Parrish JA. Photosynthesis of previtamin D3 in human and the physiologic consequences. Science. 1980;210:203–205. [PubMed: 6251551]
- 12.
- Holick MF, Richtand NM, McNeill SC, Holick SA, Frommer JE, Henley JW, Potts JT Jr. Isolation and identification of previtamin D3 from the skin of rats exposed to ultraviolet irradiation. Biochemistry. 1979;18(6):1003–1008. [PubMed: 218609]
- 13.
- Webb AR, DeCosta BR, Holick MF. Sunlight regulates the cutaneous production of vitamin D3 by causing its photodegradation. Journal of Clinical Endocrinology and Metabolism. 1989;68(5):882–887. [PubMed: 2541158]
- 14.
- Bell NH, Greene A, Epstein S, Oexmann MJ, Shaw S, Shary J. Evidence for alteration of the vitamin D-endocrine system in blacks. Journal of Clinical Investigation. 1985;76(2):470–473. [PMC free article: PMC423843] [PubMed: 3839801]
- 15.
- Webb AR, Kline L, Holick MF. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. Journal of Clinical Endocrinology and Metabolism. 1988;67(2):373–378. [PubMed: 2839537]
- 16.
- Matsuoka LY, Wortsman J, Dannenberg MJ, Hollis BW, Lu Z, Holick MF. Clothing prevents ultraviolet-B radiation-dependent photosynthesis of vitamin D3. The Journal of clinical endocrinology and metabolism. 1992;75(4):1099–1103. [PubMed: 1328275]
- 17.
- Matsuoka LY, Ide L, Wortsman J, MacLaughlin JA, Holick MF. Sunscreens suppress cutaneous vitamin D3 synthesis. Journal of Clinical Endocrinology and Metabolism. 1987;64(6):1165–1168. [PubMed: 3033008]
- 18.
- Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(39):15650–15655. [PMC free article: PMC3785760] [PubMed: 24019477]
- 19.
- Thacher TD, Fischer PR, Singh RJ, Roizen J, Levine MA. CYP2R1 Mutations Impair Generation of 25-hydroxyvitamin D and Cause an Atypical Form of Vitamin D Deficiency. The Journal of clinical endocrinology and metabolism. 2015;100(7):E1005–1013. [PMC free article: PMC4490307] [PubMed: 25942481]
- 20.
- Andersson S, Davis DL, Dahlbäck H, Jörnvall H, Russell DW. Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. Journal of Biological Chemistry. 1989;264(14):8222–8229. [PubMed: 2722778]
- 21.
- Usui E, Noshiro M, Okuda K. Molecular cloning of cDNA for vitamin D3 25-hydroxylase from rat liver mitochondria. Febs Letters. 1990;262(1):135–138. [PubMed: 2318307]
- 22.
- Cali JJ, Russell DW. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. Journal of Biological Chemistry. 1991;266(12):7774–7778. [PubMed: 1708392]
- 23.
- Ichikawa F, Sato K, Nanjo M, Nishii Y, Shinki T, Takahashi N, Suda T. Mouse primary osteoblasts express vitamin D3 25-hydroxylase mRNA and convert 1 alpha-hydroxyvitamin D3 into 1 alpha,25-dihydroxyvitamin D3. Bone. 1995;16(1):129–135. [PubMed: 7742071]
- 24.
- Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. Journal of Biological Chemistry. 1991;266(12):7779–7783. [PMC free article: PMC4449724] [PubMed: 2019602]
- 25.
- Leitersdorf E, Reshef A, Meiner V, Levitzki R, Schwartz SP, Dann EJ, Berkman N, Cali JJ, Klapholz L, Berginer VM. Frameshift and splice-junction mutations in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis in Jews or Moroccan origin. Journal of Clinical Investigation. 1993;91(6):2488–2496. [PMC free article: PMC443309] [PubMed: 8514861]
- 26.
- Berginer VM, Shany S, Alkalay D, Berginer J, Dekel S, Salen G, Tint GS, Gazit D. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism: Clinical and Experimental. 1993;42(1):69–74. [see comments] [PubMed: 8446051]
- 27.
- Leitersdorf E, Safadi R, Meiner V, Reshef A, Björkhem I, Friedlander Y, Morkos S, Berginer VM. Cerebrotendinous xanthomatosis in the Israeli Druze: molecular genetics and phenotypic characteristics. American Journal of Human Genetics. 1994;55(5):907–915. [PMC free article: PMC1918342] [PubMed: 7977352]
- 28.
- Guo YD, Strugnell S, Jones G. Identification of a human liver mitochondrial cytochrome P-450 cDNA corresponding to the vitamin D3-25-hydroxylase. Journal of Bone and Mineral Research. 1991;6:S120.
- 29.
- Guo YD, Strugnell S, Back DW, Jones G. Transfected human liver cytochrome P-450 hydroxylates vitamin D analogs at different side-chain positions. Proc Natl Acad Sci U S A. 1993;90(18):8668–8672. [PMC free article: PMC47419] [PubMed: 7690968]
- 30.
- Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. The Journal of biological chemistry. 2003;278(39):38084–38093. [PMC free article: PMC4450819] [PubMed: 12867411]
- 31.
- Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(20):7711–7715. [PMC free article: PMC419671] [PubMed: 15128933]
- 32.
- Theodoropoulos C, Demers C, Mirshahi A, Gascon-Barre M. 1,25-Dihydroxyvitamin D(3) downregulates the rat intestinal vitamin D(3)-25-hydroxylase CYP27A. Am J Physiol Endocrinol Metab. 2001;281(2):E315–325. [PubMed: 11440908]
- 33.
- Axen E, Postlind H, Wikvall K. Effects on CYP27 mRNA expression in rat kidney and liver by 1 alpha, 25- dihydroxyvitamin D3, a suppressor of renal 25-hydroxyvitamin D3 1 alpha- hydroxylase activity. Biochem Biophys Res Commun. 1995;215(1):136–141. [PubMed: 7575580]
- 34.
- Vlahcevic ZR, Jairath SK, Heuman DM, Stravitz RT, Hylemon PB, Avadhani NG, Pandak WM. Transcriptional regulation of hepatic sterol 27-hydroxylase by bile acids. Am J Physiol. 1996;270(4 Pt 1):G646–652. [PubMed: 8928794]
- 35.
- Twisk J, Hoekman MF, Lehmann EM, Meijer P, Mager WH, Princen HM. Insulin suppresses bile acid synthesis in cultured rat hepatocytes by down-regulation of cholesterol 7 alpha-hydroxylase and sterol 27- hydroxylase gene transcription. Hepatology. 1995;21(2):501–510. [PubMed: 7843724]
- 36.
- Stravitz RT, Vlahcevic ZR, Russell TL, Heizer ML, Avadhani NG, Hylemon PB. Regulation of sterol 27-hydroxylase and an alternative pathway of bile acid biosynthesis in primary cultures of rat hepatocytes. J Steroid Biochem Mol Biol. 1996;57(5-6):337–347. [PubMed: 8639470]
- 37.
- Roizen JD, Long C, Casella A, O'Lear L, Caplan I, Lai M, Sasson I, Singh R, Makowski AJ, Simmons R, Levine MA. Obesity Decreases Hepatic 25-Hydroxylase Activity Causing Low Serum 25-Hydroxyvitamin D. J Bone Miner Res. 2019:e3686. [PMC free article: PMC6663580] [PubMed: 30790351]
- 38.
- Aatsinki SM, Elkhwanky MS, Kummu O, Karpale M, Buler M, Viitala P, Rinne V, Mutikainen M, Tavi P, Franko A, Wiesner RJ, Chambers KT, Finck BN, Hakkola J. Fasting-Induced Transcription Factors Repress Vitamin D Bioactivation, a Mechanism for Vitamin D Deficiency in Diabetes. Diabetes. 2019;68(5):918–931. [PMC free article: PMC6477896] [PubMed: 30833469]
- 39.
- Saarem K, Pedersen JI. Sex differences in the hydroxylation of cholecalciferol and of 5 beta-cholestane-3 alpha, 7 alpha, 12 alpha-triol in rat liver. Biochemical Journal. 1987;247(1):73–78. [PMC free article: PMC1148371] [PubMed: 2825658]
- 40.
- Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, Portale AA. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961–1970. [PubMed: 9415400]
- 41.
- Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277(5333):1827–1830. [PubMed: 9295274]
- 42.
- St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH. The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1997;12(10):1552-1559. [PubMed: 9333115]
- 43.
- Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca HF, Suda T. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci U S A. 1997;94(24):12920–12925. [PMC free article: PMC24239] [PubMed: 9371776]
- 44.
- Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338(10):653–661. [PubMed: 9486994]
- 45.
- Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, Portale AA, Miller WL. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63(6):1694–1702. [PMC free article: PMC1377641] [PubMed: 9837822]
- 46.
- Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)- hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D- deficiency rickets. Endocrinology. 2001;142(7):3135–3141. [PubMed: 11416036]
- 47.
- Bikle DD, Nemanic MK, Whitney JO, Elias PW. Neonatal human foreskin keratinocytes produce 1,25-dihydroxyvitamin D3. Biochemistry. 1986;25(7):1545–1548. [PubMed: 2423114]
- 48.
- Murayama A, Takeyama K, Kitanaka S, Kodera Y, Kawaguchi Y, Hosoya T, Kato S. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology. 1999;140(5):2224–2231. [PubMed: 10218975]
- 49.
- Panda DK, Al Kawas S, Seldin MF, Hendy GN, Goltzman D. 25-hydroxyvitamin D 1alpha-hydroxylase: structure of the mouse gene, chromosomal assignment, and developmental expression. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2001;16(1):46-56. [PubMed: 11149489]
- 50.
- Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. The Journal of clinical endocrinology and metabolism. 2001;86(2):888–894. [PubMed: 11158062]
- 51.
- Bikle DD. Extra renal synthesis of 1,25 dihydroxyvitamin D and its Health Implications. Clin Rev in Bone and Min Metab. 2009;7:114–125.
- 52.
- Bikle DD, Pillai S. Vitamin D, calcium, and epidermal differentiation. Endocrine Review. 1993;14:3–19. [PubMed: 8491153]
- 53.
- Horiuchi N, Suda T, Takahashi H, Shimazawa E, Ogata E. In vivo evidence for the intermediary role of 3',5'-cyclic AMP in parathyroid hormone-induced stimulation of 1alpha,25-dihydroxyvitamin D3 synthesis in rats. Endocrinology. 1977;101(3):969–974. [PubMed: 196836]
- 54.
- Rasmussen H, Wong M, Bikle D, Goodman DB. Hormonal control of the renal conversion of 25-hydroxycholecalciferol to 1,25-dihydroxycholecalciferol. The Journal of clinical investigation. 1972;51(9):2502–2504. [PMC free article: PMC292420] [PubMed: 4344734]
- 55.
- Rost CR, Bikle DD, Kaplan RA. In vitro stimulation of 25-hydroxycholecalciferol 1 alpha-hydroxylation by parathyroid hormone in chick kidney slices: evidence for a role for adenosine 3',5'-monophosphate. Endocrinology. 1981;108(3):1002–1006. [PubMed: 6257493]
- 56.
- Henry HL. Parathyroid hormone modulation of 25-hydroxyvitamin D3 metabolism by cultured chick kidney cells is mimicked and enhanced by forskolin. Endocrinology. 1985;116(2):503–510. [PubMed: 2981664]
- 57.
- Armbrecht HJ, Forte LR, Wongsurawat N, Zenser TV, Davis BB. Forskolin increases 1,25-dihydroxyvitamin D3 production by rat renal slices in vitro. Endocrinology. 1984;114(2):644–649. [PubMed: 6317364]
- 58.
- Janulis M, Tembe V, Favus MJ. Role of protein kinase C in parathyroid hormone stimulation of renal 1,25-dihydroxyvitamin D3 secretion. The Journal of clinical investigation. 1992;90(6):2278–2283. [PMC free article: PMC443379] [PubMed: 1334973]
- 59.
- Janulis M, Wong MS, Favus MJ. Structure-function requirements of parathyroid hormone for stimulation of 1,25-dihydroxyvitamin D3 production by rat renal proximal tubules. Endocrinology. 1993;133(2):713–719. [PubMed: 8344210]
- 60.
- Brenza HL, Kimmel-Jehan C, Jehan F, Shinki T, Wakino S, Anazawa H, Suda T, DeLuca HF. Parathyroid hormone activation of the 25-hydroxyvitamin D3-1alpha- hydroxylase gene promoter. Proc Natl Acad Sci U S A. 1998;95(4):1387–1391. [PMC free article: PMC19012] [PubMed: 9465024]
- 61.
- Zierold C, Nehring JA, DeLuca HF. Nuclear receptor 4A2 and C/EBPbeta regulate the parathyroid hormone-mediated transcriptional regulation of the 25-hydroxyvitamin D3-1alpha-hydroxylase. Archives of biochemistry and biophysics. 2007;460(2):233–239. [PubMed: 17224121]
- 62.
- Takeyama K, Kato S. The vitamin D3 1alpha-hydroxylase gene and its regulation by active vitamin D3. Biosci Biotechnol Biochem. 2011;75(2):208–213. [PubMed: 21307571]
- 63.
- Bikle DD, Murphy EW, Rasmussen H. The ionic control of 1,25-dihydroxyvitamin D3 synthesis in isolated chick renal mitochondria. The role of calcium as influenced by inorganic phosphate and hydrogen-ion. The Journal of clinical investigation. 1975;55(2):299–304. [PMC free article: PMC301748] [PubMed: 236327]
- 64.
- Bushinsky DA, Riera GS, Favus MJ, Coe FL. Evidence that blood ionized calcium can regulate serum 1,25(OH)2D3 independently of parathyroid hormone and phosphorus in the rat. The Journal of clinical investigation. 1985;76(4):1599–1604. [PMC free article: PMC424140] [PubMed: 3840495]
- 65.
- Hulter HN, Halloran BP, Toto RD, Peterson JC. Long-term control of plasma calcitriol concentration in dogs and humans. Dominant role of plasma calcium concentration in experimental hyperparathyroidism. The Journal of clinical investigation. 1985;76(2):695–702. [PMC free article: PMC423880] [PubMed: 3928683]
- 66.
- Hughes MR, Brumbaugh PF, Hussler MR, Wergedal JE, Baylink DJ. Regulation of serum 1alpha,25-dihydroxyvitamin D3 by calcium and phosphate in the rat. Science. 1975;190(4214):578–580. [PubMed: 1188357]
- 67.
- Portale AA, Halloran BP, Morris RC Jr. Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. The Journal of clinical investigation. 1987;80(4):1147–1154. [PMC free article: PMC442358] [PubMed: 3654974]
- 68.
- Condamine L, Menaa C, Vrtovsnik F, Vztovsnik F, Friedlander G, Garabedian M. Local action of phosphate depletion and insulin-like growth factor 1 on in vitro production of 1,25-dihydroxyvitamin D by cultured mammalian kidney cells. The Journal of clinical investigation. 1994;94(4):1673–1679. [PMC free article: PMC295330] [PubMed: 7929846]
- 69.
- Gray RW. Control of plasma 1,25-(OH)2-vitamin D concentrations by calcium and phosphorus in the rat: effects of hypophysectomy. Calcif Tissue Int. 1981;33(5):485–488. [PubMed: 6797701]
- 70.
- Yoshida T, Yoshida N, Monkawa T, Hayashi M, Saruta T. Dietary phosphorus deprivation induces 25-hydroxyvitamin D(3) 1alpha- hydroxylase gene expression. Endocrinology. 2001;142(5):1720–1726. [PubMed: 11316734]
- 71.
- Gray RW, Garthwaite TL, Phillips LS. Growth hormone and triiodothyronine permit an increase in plasma 1,25(OH)2D concentrations in response to dietary phosphate deprivation in hypophysectomized rats. Calcif Tissue Int. 1983;35(1):100–106. [PubMed: 6839183]
- 72.
- Halloran BP, Spencer EM. Dietary phosphorus and 1,25-dihydroxyvitamin D metabolism: influence of insulin-like growth factor I. Endocrinology. 1988;123(3):1225–1229. [PubMed: 3402383]
- 73.
- Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. The Journal of biological chemistry. 2003;278(4):2206–2211. [PubMed: 12419819]
- 74.
- White KE, Evans WE, O'Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature Genetics. 2000;26:345. [PubMed: 11062477]
- 75.
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(11):6500–6505. [PMC free article: PMC33497] [PubMed: 11344269]
- 76.
- Blau JE, Collins MT. The PTH-Vitamin D-FGF23 axis. Reviews in endocrine & metabolic disorders. 2015;16(2):165–174. [PubMed: 26296372]
- 77.
- Meyer MB, Benkusky NA, Kaufmann M, Lee SM, Redfield RR, Jones G, Pike JW. Targeted genomic deletions identify diverse enhancer functions and generate a kidney-specific, endocrine-deficient Cyp27b1 pseudo-null mouse. The Journal of biological chemistry. 2019;294(24):9518–9535. [PMC free article: PMC6579472] [PubMed: 31053643]
- 78.
- Schuster I, Egger H, Astecker N, Herzig G, Schussler M, Vorisek G. Selective inhibitors of CYP24: mechanistic tools to explore vitamin D metabolism in human keratinocytes. Steroids. 2001;66(3-5):451–462. [PubMed: 11179754]
- 79.
- Xie Z, Munson S, Huang N, Schuster I, Portale AA, Miller WL, Bikle DD. The mechanism of 1,25-Dihydroxyvitamin D3 auto-regulation in keratinocytes. j bone min Res (Program & abstracts). 2001;16 suppl 1:S556.
- 80.
- Kato S, Fujiki R, Kim MS, Kitagawa H. Ligand-induced transrepressive function of VDR requires a chromatin remodeling complex, WINAC. The Journal of steroid biochemistry and molecular biology. 2007;103(3-5):372–380. [PubMed: 17368181]
- 81.
- Akiyoshi-Shibata M, Sakaki T, Ohyama Y, Noshiro M, Okuda K, Yabusaki Y. Further oxidation of hydroxycalcidiol by calcidiol 24-hydroxylase. A study with the mature enzyme expressed in Escherichia coli. Eur J Biochem. 1994;224(2):335–343. [PubMed: 7925346]
- 82.
- Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. 2012;523(1):9–18. [PubMed: 22100522]
- 83.
- Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, Misselwitz J, Klaus G, Kuwertz-Broking E, Fehrenbach H, Wingen AM, Guran T, Hoenderop JG, Bindels RJ, Prosser DE, Jones G, Konrad M. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(5):410–421. [PubMed: 21675912]
- 84.
- Shah AD, Hsiao EC, O'Donnell B, Salmeen K, Nussbaum R, Krebs M, Baumgartner-Parzer S, Kaufmann M, Jones G, Bikle DD, Wang Y, Mathew AS, Shoback D, Block-Kurbisch I. Maternal Hypercalcemia Due to Failure of 1,25-Dihydroxyvitamin-D3 Catabolism in a Patient With CYP24A1 Mutations. The Journal of clinical endocrinology and metabolism. 2015;100(8):2832–2836. [PMC free article: PMC4524985] [PubMed: 26097993]
- 85.
- St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, Depovere J, Mathieu C, Christakos S, Demay MB, Glorieux FH. Deficient mineralization of intramembranous bone in vitamin D-24- hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141(7):2658–2666. [PubMed: 10875271]
- 86.
- Hahn CN, Kerry DM, Omdahl JL, May BK. Identification of a vitamin D responsive element in the promoter of the rat cytochrome P450(24) gene. Nucleic Acids Res. 1994;22(12):2410–2416. [PMC free article: PMC523703] [PubMed: 8036172]
- 87.
- Chen KS, DeLuca HF. Cloning of the human 1 alpha,25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim Biophys Acta. 1995;1263(1):1–9. [PubMed: 7632726]
- 88.
- Ohyama Y, Ozono K, Uchida M, Shinki T, Kato S, Suda T, Yamamoto O, Noshiro M, Kato Y. Identification of a vitamin D-responsive element in the 5'-flanking region of the rat 25-hydroxyvitamin D3 24-hydroxylase gene. The Journal of biological chemistry. 1994;269(14):10545–10550. [PubMed: 8144641]
- 89.
- Chen ML, Boltz MA, Armbrecht HJ. Effects of 1,25-dihydroxyvitamin D3 and phorbol ester on 25- hydroxyvitamin D3 24-hydroxylase cytochrome P450 messenger ribonucleic acid levels in primary cultures of rat renal cells. Endocrinology. 1993;132(4):1782–1788. [PubMed: 7681765]
- 90.
- Pike JW, Kerner SA, Jin CH, Allegretto EA, Elgort M. Direct activation of the human 25-(OH)2D3 and PMA: Identification of cis elements and transactivators. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1995;10:S144.
- 91.
- Garabedian M, Holick MF, Deluca HF, Boyle IT. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc Natl Acad Sci U S A. 1972;69(7):1673–1676. [PMC free article: PMC426775] [PubMed: 4340153]
- 92.
- Shigematsu T, Horiuchi N, Ogura Y, Miyahara T, Suda T. Human parathyroid hormone inhibits renal 24-hydroxylase activity of 25- hydroxyvitamin D3 by a mechanism involving adenosine 3',5'- monophosphate in rats. Endocrinology. 1986;118(4):1583–1589. [PubMed: 3004922]
- 93.
- Armbrecht HJ, Wongsurawat N, Zenser TV, Davis BB. Effect of PTH and 1,25(OH)2D3 on renal 25(OH)D3 metabolism, adenylate cyclase, and protein kinase. Am J Physiol. 1984;246(1 Pt 1):E102–107. [PubMed: 6320659]
- 94.
- Armbrecht HJ, Hodam TL, Boltz MA, Partridge NC, Brown AJ, Kumar VB. Induction of the vitamin D 24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 is regulated by parathyroid hormone in UMR106 osteoblastic cells. Endocrinology. 1998;139(8):3375–3381. [PubMed: 9681485]
- 95.
- Armbrecht HJ, Wongsurawat VJ, Hodam TL, Wongsurawat N. Insulin markedly potentiates the capacity of parathyroid hormone to increase expression of 25-hydroxyvitamin D3-24-hydroxylase in rat osteoblastic cells in the presence of 1,25-dihydroxyvitamin D3. FEBS Lett. 1996;393(1):77–80. [PubMed: 8804428]
- 96.
- Meyer MB, Lee SM, Carlson AH, Benkusky NA, Kaufmann M, Jones G, Pike JW. A chromatin-based mechanism controls differential regulation of the cytochrome P450 gene Cyp24a1 in renal and non-renal tissues. The Journal of biological chemistry. 2019;294(39):14467–14481. [PMC free article: PMC6768633] [PubMed: 31439663]
- 97.
- Inoue Y, Segawa H, Kaneko I, Yamanaka S, Kusano K, Kawakami E, Furutani J, Ito M, Kuwahata M, Saito H, Fukushima N, Kato S, Kanayama HO, Miyamoto K. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005;390(Pt 1):325–331. [PMC free article: PMC1184586] [PubMed: 15885032]
- 98.
- Wu S, Grieff M, Brown AJ. Regulation of renal vitamin D-24-hydroxylase by phosphate: effects of hypophysectomy, growth hormone and insulin-like growth factor I. Biochem Biophys Res Commun. 1997;233(3):813–817. [PubMed: 9168939]
- 99.
- Cooke NE, Haddad JG. Vitamin D binding protein (Gc-globulin). Endocrine reviews. 1989;10(3):294–307. [PubMed: 2476303]
- 100.
- Bikle DD, Gee E, Halloran B, Haddad JG. Free 1,25-dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. The Journal of clinical investigation. 1984;74(6):1966–1971. [PMC free article: PMC425383] [PubMed: 6549014]
- 101.
- Bikle DD, Siiteri PK, Ryzen E, Haddad JG. Serum protein binding of 1,25-dihydroxyvitamin D: a reevaluation by direct measurement of free metabolite levels. The Journal of clinical endocrinology and metabolism. 1985;61(5):969–975. [PubMed: 3840175]
- 102.
- Bikle DD, Gee E, Halloran B, Kowalski MA, Ryzen E, Haddad JG. Assessment of the free fraction of 25-hydroxyvitamin D in serum and its regulation by albumin and the vitamin D-binding protein. The Journal of clinical endocrinology and metabolism. 1986;63(4):954–959. [PubMed: 3745408]
- 103.
- Bikle DD, Halloran BP, Gee E, Ryzen E, Haddad JG. Free 25-hydroxyvitamin D levels are normal in subjects with liver disease and reduced total 25-hydroxyvitamin D levels. The Journal of clinical investigation. 1986;78(3):748–752. [PMC free article: PMC423667] [PubMed: 3745436]
- 104.
- Madden K, Feldman HA, Chun RF, Smith EM, Sullivan RM, Agan AA, Keisling SM, Panoskaltsis-Mortari A, Randolph AG. Critically Ill Children Have Low Vitamin D-Binding Protein, Influencing Bioavailability of Vitamin D. Ann Am Thorac Soc. 2015;12(11):1654–1661. [PMC free article: PMC5467088] [PubMed: 26356094]
- 105.
- Nielson CM, Jones KS, Chun RF, Jacobs JM, Wang Y, Hewison M, Adams JS, Swanson CM, Lee CG, Vanderschueren D, Pauwels S, Prentice A, Smith RD, Shi T, Gao Y, Schepmoes AA, Zmuda JM, Lapidus J, Cauley JA, Bouillon R, Schoenmakers I, Orwoll ES. Osteoporotic Fractures in Men Research G. Free 25-Hydroxyvitamin D: Impact of Vitamin D Binding Protein Assays on Racial-Genotypic Associations. The Journal of clinical endocrinology and metabolism. 2016;101(5):2226–2234. [PMC free article: PMC4870848] [PubMed: 27007693]
- 106.
- Pettifor JM, Bikle DD, Cavaleros M, Zachen D, Kamdar MC, Ross FP. Serum levels of free 1,25-dihydroxyvitamin D in vitamin D toxicity. Ann Intern Med. 1995;122(7):511–513. [PubMed: 7872586]
- 107.
- Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, Liebhaber SA, Cooke NE. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. The Journal of clinical investigation. 1999;103(2):239–251. [PMC free article: PMC407885] [PubMed: 9916136]
- 108.
- Zella LA, Shevde NK, Hollis BW, Cooke NE, Pike JW. Vitamin D-binding protein influences total circulating levels of 1,25-dihydroxyvitamin D3 but does not directly modulate the bioactive levels of the hormone in vivo. Endocrinology. 2008;149(7):3656–3667. [PMC free article: PMC2453093] [PubMed: 18372326]
- 109.
- Henderson CM, Fink SL, Bassyouni H, Argiropoulos B, Brown L, Laha TJ, Jackson KJ, Lewkonia R, Ferreira P, Hoofnagle AN, Marcadier JL. Vitamin D-Binding Protein Deficiency and Homozygous Deletion of the GC Gene. The New England journal of medicine. 2019;380(12):1150–1157. [PMC free article: PMC7898410] [PubMed: 30893535]
- 110.
- Bikle D, Bouillon R, Thadhani R, Schoenmakers I. Vitamin D metabolites in captivity? Should we measure free or total 25(OH)D to assess vitamin D status? The Journal of steroid biochemistry and molecular biology. 2017;173:105–116. [PMC free article: PMC9005158] [PubMed: 28093353]
- 111.
- Malmstroem S, Rejnmark L, Imboden JB, Shoback DM, Bikle DD. Current Assays to Determine Free 25-Hydroxyvitamin D in Serum. J AOAC Int. 2017;100:1323–1327. [PubMed: 28492133]
- 112.
- Nykjaer A, Dragun D, Walther D, Vorum H, Jacobsen C, Herz J, Melsen F, Christensen EI, Willnow TE. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96(4):507–515. [PubMed: 10052453]
- 113.
- Arnaud J, Constans J. Affinity differences for vitamin D metabolites associated with the genetic isoforms of the human serum carrier protein (DBP). Hum Genet. 1993;92(2):183–188. [PubMed: 8370586]
- 114.
- Cooke NE, David EV. Serum vitamin D-binding protein is a third member of the albumin and alpha fetoprotein gene family. The Journal of clinical investigation. 1985;76(6):2420–2424. [PMC free article: PMC424397] [PubMed: 2416779]
- 115.
- Cooke NE, Levan G, Szpirer J. The rat vitamin D binding protein (Gc-globulin) gene is syntenic with the rat albumin and alpha-fetoprotein genes on chromosome 14. Cytogenet Cell Genet. 1987;44(2-3):98–100. [PubMed: 2436858]
- 116.
- Guha C, Osawa M, Werner PA, Galbraith RM, Paddock GV. Regulation of human Gc (vitamin D--binding) protein levels: hormonal and cytokine control of gene expression in vitro. Hepatology. 1995;21(6):1675–1681. [PubMed: 7539397]
- 117.
- Lees A, Haddad JG, Lin S. Brevin and DBP comparison of the effects of two serum protein on actin assembly and disassembly. Biochemistry. 1984;23:3038–3047. [PubMed: 6547850]
- 118.
- Yamamoto N, Homma S, Haddad JG, Kowalski MA. Vitamin D3 binding protein required for in vitro activation of macrophages after alkylglycerol treatment of mouse peritoneal cells. Immunology. 1991;74(3):420–424. [PMC free article: PMC1384634] [PubMed: 1769691]
- 119.
- Schneider GB, Benis KA, Flay NW, Ireland RA, Popoff SN. Effects of vitamin D binding protein-macrophage activating factor (DBP- MAF) infusion on bone resorption in two osteopetrotic mutations. Bone. 1995;16(6):657–662. [PubMed: 7669443]
- 120.
- Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, Liebhaber SA, Cooke NE. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. The Journal of clinical investigation. 1999;103(2):239–251. [PMC free article: PMC407885] [PubMed: 9916136]
- 121.
- Haussler MR, Norman AW. Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci U S A. 1969;62(1):155–162. [PMC free article: PMC285968] [PubMed: 5253652]
- 122.
- McDonnell DP, Mangelsdorf DJ, Pike JW, Haussler MR, O'Malley BW. Molecular cloning of complementary DNA encoding the avian receptor for vitamin D. Science. 1987;235(4793):1214–1217. [PubMed: 3029866]
- 123.
- Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, Shine J, O'Malley BW. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85(10):3294–3298. [PMC free article: PMC280195] [PubMed: 2835767]
- 124.
- Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242(4886):1702–1705. [PubMed: 2849209]
- 125.
- Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, Masushige S, Fukamizu A, Matsumoto T, Kato S. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16(4):391–396. [PubMed: 9241280]
- 126.
- Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(18):9831–9835. [PMC free article: PMC23277] [PubMed: 9275211]
- 127.
- Walters MR. Newly identified actions of the vitamin D endocrine system. Endocr Rev. 1992;13(4):719–764. [PubMed: 1333949]
- 128.
- Harris SS, Eccleshall TR, Gross C, Dawson-Hughes B, Feldman D. The vitamin D receptor start codon polymorphism (FokI) and bone mineral density in premenopausal American black and white women. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1997;12(7):1043-1048. [PubMed: 9200003]
- 129.
- Rachez C, Gamble M, Chang CP, Atkins GB, Lazar MA, Freedman LP. The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Mol Cell Biol. 2000;20(8):2718–2726. [PMC free article: PMC85487] [PubMed: 10733574]
- 130.
- Makowski A, Brzostek S, Cohen RN, Hollenberg AN. Determination of nuclear receptor corepressor interactions with the thyroid hormone receptor. Molecular endocrinology (Baltimore, Md). 2003;17(2):273–286. [PubMed: 12554754]
- 131.
- Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173–179. [PubMed: 10678179]
- 132.
- Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. The EMBO journal. 2012;31(2):291–300. [PMC free article: PMC3261568] [PubMed: 22179700]
- 133.
- Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3(1):87–94. [PubMed: 8548460]
- 134.
- Chen S, Cui J, Nakamura K, Ribeiro RC, West BL, Gardner DG. Coactivator-vitamin D receptor interactions mediate inhibition of the atrial natriuretic peptide promoter. The Journal of biological chemistry. 275(20):15039–15048. [PubMed: 10809746]
- 135.
- McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20(3):321–344. [PubMed: 10368774]
- 136.
- Polly P, Herdick M, Moehren U, Baniahmad A, Heinzel T, Carlberg C. VDR-Alien: a novel, DNA-selective vitamin D(3) receptor-corepressor partnership. Faseb J. 2000;14(10):1455–1463. [PubMed: 10877839]
- 137.
- Xie Z, Chang S, Oda Y, Bikle DD. Hairless suppresses vitamin D receptor transactivation in human keratinocytes. Endocrinology. 2006;147(1):314–323. [PubMed: 16269453]
- 138.
- Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. The Journal of biological chemistry. 2003;278(40):38665–38674. [PubMed: 12847098]
- 139.
- Bikle DD, Elalieh H, Chang S, Xie Z, Sundberg JP. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol. 2006;207:340–353. [PubMed: 16419036]
- 140.
- Leo C, Chen JD. The SRC family of nuclear receptor coactivators. Genes. 2000;245:1–11. [PubMed: 10713439]
- 141.
- Rachez C, Lemon BD, Suldan Z, Bromleigh V, Gamble M, Naar AM, Erdjument-Bromage H, Tempst P, Freedman LP. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature. 1999;398(6730):824–828. [PubMed: 10235266]
- 142.
- Teichert A, Arnold LA, Otieno S, Oda Y, Augustinaite I, Geistlinger TR, Kriwacki RW, Guy RK, Bikle DD. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry. 2009;48(7):1454–1461. [PMC free article: PMC2654718] [PubMed: 19183053]
- 143.
- Oda Y. Journal of Investigative Dermatology. 2001:116. Abstract 809.
- 144.
- Hawker NP, Pennypacker SD, Chang SM, Bikle DD. Regulation of Human Epidermal Keratinocyte Differentiation by the Vitamin D Receptor and its Coactivators DRIP205, SRC2, and SRC3. The Journal of investigative dermatology. 2007;127:874. [PubMed: 17082781]
- 145.
- Schauber J, Oda Y, Buchau AS, Yun QC, Steinmeyer A, Zugel U, Bikle DD, Gallo RL. Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1,25-dihydroxyvitamin D3. J Invest Dermatol. 2008;128(4):816–824. [PubMed: 17943182]
- 146.
- Oda Y, Uchida Y, Moradian S, Crumrine D, Elias P, Bikle D. Vitamin D receptor and coactivators SRC 2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol. 2009;129(6):1367–1378. [PMC free article: PMC2843519] [PubMed: 19052561]
- 147.
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. [PMC free article: PMC3653129] [PubMed: 23582322]
- 148.
- Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa M, Watanabe M, Kashiwagi K, Toriyabe T, Kawabata M, Miyazono K, Kato S. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283(5406):1317–1321. [PubMed: 10037600]
- 149.
- Hsieh JC, Jurutka PW, Nakajima S, Galligan MA, Haussler CA, Shimizu Y, Shimizu N, Whitfield GK, Haussler MR. Phosphorylation of the human vitamin D receptor by protein kinase C. Biochemical and functional evaluation of the serine 51 recognition site. The Journal of biological chemistry. 1993;268(20):15118–15126. [PubMed: 8392065]
- 150.
- Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de Herreros AG, Lafarga M, Munoz A. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. The Journal of cell biology. 2001;154(2):369–387. [PMC free article: PMC2150773] [PubMed: 11470825]
- 151.
- Palmer HG, Anjos-Afonso F, Carmeliet G, Takeda H, Watt FM. The Vitamin D Receptor Is a Wnt Effector that Controls Hair Follicle Differentiation and Specifies Tumor Type in Adult Epidermis. PloS one. 2008;3(1):e1483. [PMC free article: PMC2198947] [PubMed: 18213391]
- 152.
- Kurokawa R, Yu VC, Naar A, Kyakumoto S, Han Z, Silverman S, Rosenfeld MG, Glass CK. Differential orientations of the DNA-binding domain and carboxy- terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes Dev. 1993;7(7B):1423–1435. [PubMed: 8392479]
- 153.
- Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. The Journal of biological chemistry. 2000;275(52):41114–41123. [PubMed: 11001945]
- 154.
- Xie Z, Bikle DD. Cloning of the human phospholipase C-gamma1 promoter and identification of a DR6-type vitamin D-responsive element. The Journal of biological chemistry. 1997;272(10):6573–6577. [PubMed: 9045685]
- 155.
- Schrader M, Muller KM, Nayeri S, Kahlen JP, Carlberg C. Vitamin D3-thyroid hormone receptor heterodimer polarity directs ligand sensitivity of transactivation. Nature. 1994;370(6488):382–386. [PubMed: 8047145]
- 156.
- Lemon BD, Freedman LP. Selective effects of ligands on vitamin D3 receptor- and retinoid X receptor-mediated gene activation in vivo. Mol Cell Biol. 1996;16(3):1006–1016. [PMC free article: PMC231083] [PubMed: 8622645]
- 157.
- Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D3. Rheumatic diseases clinics of North America. 2012;38(1):13–27. [PMC free article: PMC4448919] [PubMed: 22525840]
- 158.
- Carlberg C, Seuter S, Heikkinen S. The first genome-wide view of vitamin D receptor locations and their mechanistic implications. Anticancer research. 2012;32(1):271–282. [PubMed: 22213316]
- 159.
- MacDonald PN, Dowd DR, Nakajima S, Galligan MA, Reeder MC, Haussler CA, Ozato K, Haussler MR. Retinoid X receptors stimulate and 9-cis retinoic acid inhibits 1,25- dihydroxyvitamin D3-activated expression of the rat osteocalcin gene. Mol Cell Biol. 1993;13(9):5907–5917. [PMC free article: PMC360339] [PubMed: 8395017]
- 160.
- Kang S, Li XY, Duell EA, Voorhees JJ. The retinoid X receptor agonist 9-cis-retinoic acid and the 24- hydroxylase inhibitor ketoconazole increase activity of 1,25- dihydroxyvitamin D3 in human skin in vivo. The Journal of investigative dermatology. 1997;108(4):513–518. [PubMed: 9077483]
- 161.
- Gill RK, Christakos S. Identification of sequence elements in mouse calbindin-D28k gene that confer 1,25-dihydroxyvitamin D3- and butyrate-inducible responses. Proc Natl Acad Sci U S A. 1993;90(7):2984–2988. [PMC free article: PMC46221] [PubMed: 8464915]
- 162.
- Liu SM, Koszewski N, Lupez M, Malluche HH, Olivera A, Russell J. Characterization of a response element in the 5'-flanking region of the avian (chicken) PTH gene that mediates negative regulation of gene transcription by 1,25-dihydroxyvitamin D3 and binds the vitamin D3 receptor. Mol Endocrinol. 1996;10(2):206–215. [PubMed: 8825560]
- 163.
- Kremer R, Sebag M, Champigny C, Meerovitch K, Hendy GN, White J, Goltzman D. Identification and characterization of 1,25-dihydroxyvitamin D3- responsive repressor sequences in the rat parathyroid hormone-related peptide gene. The Journal of biological chemistry. 1996;271(27):16310–16316. [PubMed: 8663213]
- 164.
- Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol. 1995;15(10):5789–5799. [PMC free article: PMC230831] [PubMed: 7565732]
- 165.
- Morley P, Whitfield JF, Vanderhyden BC, Tsang BK, Schwartz JL. A new, nongenomic estrogen action: the rapid release of intracellular calcium. Endocrinology. 1992;131(3):1305–1312. [PubMed: 1505465]
- 166.
- Wistrom CA, Meizel S. Evidence suggesting involvement of a unique human sperm steroid receptor/Cl- channel complex in the progesterone-initiated acrosome reaction. Dev Biol. 1993;159(2):679–690. [PubMed: 8405689]
- 167.
- Koenig H, Fan CC, Goldstone AD, Lu CY, Trout JJ. Polyamines mediate androgenic stimulation of calcium fluxes and membrane transport in rat heart myocytes. Circ Res. 1989;64(3):415–426. [PubMed: 2537154]
- 168.
- Orchinik M, Murray TF, Moore FL. A corticosteroid receptor in neuronal membranes. Science. 1991;252(5014):1848–1851. [PubMed: 2063198]
- 169.
- Segal J. Thyroid hormone action at the level of the plasma membrane. Thyroid. 1990;1(1):83–87. [PubMed: 2135990]
- 170.
- Caffrey JM, Farach-Carson MC. Vitamin D3 metabolites modulate dihydropyridine-sensitive calcium currents in clonal rat osteosarcoma cells. The Journal of biological chemistry. 1989;264(34):20265–20274. [PubMed: 2479647]
- 171.
- Baran DT, Sorensen AM, Honeyman TW, Ray R, Holick MF. 1 alpha,25-dihydroxyvitamin D3-induced increments in hepatocyte cytosolic calcium and lysophosphatidylinositol: inhibition by pertussis toxin and 1 beta,25-dihydroxyvitamin D3. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1990;5(5):517-524. [PubMed: 2368631]
- 172.
- Morelli S, de Boland AR, Boland RL. Generation of inositol phosphates, diacylglycerol and calcium fluxes in myoblasts treated with 1,25-dihydroxyvitamin D3. Biochem J. 1993;289(Pt 3):675–679. [PMC free article: PMC1132228] [PubMed: 8382046]
- 173.
- Wali RK, Baum CL, Sitrin MD, Brasitus TA. 1,25(OH)2 vitamin D3 stimulates membrane phosphoinositide turnover, activates protein kinase C, and increases cytosolic calcium in rat colonic epithelium. The Journal of clinical investigation. 1990;85(4):1296–1303. [PMC free article: PMC296566] [PubMed: 2156898]
- 174.
- Khare S, Bolt MJ, Wali RK, Skarosi SF, Roy HK, Niedziela S, Scaglione-Sewell B, Aquino B, Abraham C, Sitrin MD, Brasitus TA, Bissonnette M. 1,25 dihydroxyvitamin D3 stimulates phospholipase C-gamma in rat colonocytes: role of c-Src in PLC-gamma activation. The Journal of clinical investigation. 1997;99(8):1831–1841. [PMC free article: PMC508007] [PubMed: 9109427]
- 175.
- Nemere I, Yoshimoto Y, Norman AW. Calcium transport in perfused duodena from normal chicks: enhancement within fourteen minutes of exposure to 1,25-dihydroxyvitamin D3. Endocrinology. 1984;115(4):1476–1483. [PubMed: 6548181]
- 176.
- Nemere I, Norman AW. Rapid action of 1,25-dihydroxyvitamin D3 on calcium transport in perfused chick duodenum: effect of inhibitors. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1987;2(2):99-107. [PubMed: 3455164]
- 177.
- de Boland AR, Norman AW. Influx of extracellular calcium mediates 1,25-dihydroxyvitamin D3- dependent transcaltachia (the rapid stimulation of duodenal Ca2+ transport). Endocrinology. 1990;127(5):2475–2480. [PubMed: 2121464]
- 178.
- de Boland AR, Norman A. Evidence for involvement of protein kinase C and cyclic adenosine 3',5' monophosphate-dependent protein kinase in the 1,25-dihydroxy-vitamin D3- mediated rapid stimulation of intestinal calcium transport, (transcaltachia). Endocrinology. 1990;127(1):39–45. [PubMed: 2163320]
- 179.
- de Boland AR, Nemere I, Norman AW. Ca2(+)-channel agonist BAY K8644 mimics 1,25(OH)2-vitamin D3 rapid enhancement of Ca2+ transport in chick perfused duodenum. Biochem Biophys Res Commun. 1990;166(1):217–222. [PubMed: 1689150]
- 180.
- Nemere I, Dormanen MC, Hammond MW, Okamura WH, Norman AW. Identification of a specific binding protein for 1 alpha,25- dihydroxyvitamin D3 in basal-lateral membranes of chick intestinal epithelium and relationship to transcaltachia. The Journal of biological chemistry. 1994;269(38):23750–23756. [PubMed: 8089147]
- 181.
- Nemere I, Safford SE, Rohe B, DeSouza MM, Farach-Carson MC. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25D3-MARRS) binding protein. The Journal of steroid biochemistry and molecular biology. 2004;89-90(1-5):281–285. [PubMed: 15225786]
- 182.
- Pedrozo HA, Schwartz Z, Rimes S, Sylvia VL, Nemere I, Posner GH, Dean DD, Boyan BD. Physiological importance of the 1,25(OH)2D3 membrane receptor and evidence for a membrane receptor specific for 24,25(OH)2D3. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1999;14(6):856-867. [PubMed: 10352093]
- 183.
- Khanal RC, Peters TM, Smith NM, Nemere I. Membrane receptor-initiated signaling in 1,25(OH)2D3-stimulated calcium uptake in intestinal epithelial cells. Journal of cellular biochemistry. 2008;105(4):1109–1116. [PubMed: 18773429]
- 184.
- Norman AW, Okamura WH, Farach-Carson MC, Allewaert K, Branisteanu D, Nemere I, Muralidharan KR, Bouillon R. Structure-function studies of 1,25-dihydroxyvitamin D3 and the vitamin D endocrine system. 1,25-dihydroxy-pentadeuterio-previtamin D3 (as a 6- s-cis analog) stimulates nongenomic but not genomic biological responses. The Journal of biological chemistry. 1993;268(19):13811–13819. [PubMed: 8390978]
- 185.
- Sequeira VB, Rybchyn MS, Tongkao-On W, Gordon-Thomson C, Malloy PJ, Nemere I, Norman AW, Reeve VE, Halliday GM, Feldman D, Mason RS. The role of the vitamin D receptor and ERp57 in photoprotection by 1alpha,25-dihydroxyvitamin D3. Molecular endocrinology (Baltimore, Md). 2012;26(4):574–582. [PMC free article: PMC3327356] [PubMed: 22322599]
- 186.
- Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(35):12876–12881. [PMC free article: PMC516488] [PubMed: 15326291]
- 187.
- Schachter D, Rosen S. Active transport of Ca45 by the small intestine and its dependence on vitamin D. Am J Physiol. 1959;196:357–362. [PubMed: 13627180]
- 188.
- Wasserman RH, Kallfelz FA, Comar CL. Active transport of calcium by rat duodenum in vivo. Science. 1961;133:883–884. [PubMed: 13783309]
- 189.
- Bikle DD. Regulation of intestinal calcium transport by vitamin D [1,25(OH)2]:role of membrane structure. In: Membrane transport and information storage. New York: Wiley-Liss. 1990, pp 191-219.
- 190.
- Wasserman RH, Fullmer CS. Vitamin D and intestinal calcium transport: facts, speculations and hypotheses. J Nutr. 1995;125(7) Suppl:1971S–1979S. [PubMed: 7602379]
- 191.
- Wongdee K, Chanpaisaeng K, Teerapornpuntakit J, Charoenphandhu N. Intestinal Calcium Absorption. Compr Physiol. 2021;11(3):2047–2073. [PubMed: 34058017]
- 192.
- Bikle DD, Shoback DM, Munson S. 1,25-dihydroxyvitamin D increases the intracellular free calcium concentration of duodenal epithelial cells. In: Vitamin D: Chemial, biochemical and clinical update. New York: Walter de Gruyter, 1985, p 416.
- 193.
- Morrissey RL, Zolock DT, Mellick PW, Bikle DD. Influence of cycloheximide and 1,25-dihydroxyvitamin D3 on mitochondrial and vesicle mineralization in the intestine. Cell Calcium. 1980;1:69–79.
- 194.
- Davis WL, Hagler HK, Jones RG, Farmer GR, Cooper OJ, Martin JH, Bridges GE, Goodman DB. Cryofixation, ultracryomicrotomy, and X-ray microanalysis of enterocytes from chick duodenum: vitamin-D-induced formation of an apical tubulovesicular system. Anat Rec. 1991;229(2):227–239. [PubMed: 2012310]
- 195.
- Nemere I, Leathers V, Norman AW. 1,25-Dihydroxyvitamin D3-mediated intestinal calcium transport. Biochemical identification of lysosomes containing calcium and calcium- binding protein (calbindin-D28K). The Journal of biological chemistry. 1986;261(34):16106–16114. [PubMed: 3023341]
- 196.
- Max EE, Goodman DB, Rasmussen H. Purification and characterization of chick intestine brush border membrane. Effects of 1alpha(OH) vitamin D3 treatment. Biochimica et biophysica acta. 1978;511(2):224–239. [PubMed: 678542]
- 197.
- Brasitus TA, Dudeja PK, Eby B, Lau K. Correction by 1,25(OH)2D3 of the abnormal fluidity and lipid composition of enterocyte brush border membranes in vitamin D-deprived rats. Journal of Biological Chemistry. 1981;256:3354–3360. [PubMed: 3782126]
- 198.
- Matsumoto T, Fontaine O, Rasmussen H. Effect of 1,25-dihydroxyvitamin D3 on phospholipid metabolism in chick duodenal mucosal cell. Relationship to its mechanism of action. The Journal of biological chemistry. 1981;256(7):3354–3360. [PubMed: 6894144]
- 199.
- Bikle DD, Whitney J, Munson S. The relationship of membrane fluidity to calcium flux in chick intestinal brush border membranes. Endocrinology. 1984;114(1):260–267. [PubMed: 6546306]
- 200.
- Peng JB, Chen XZ, Berger UV, Vassilev PM, Tsukaguchi H, Brown EM, Hediger MA. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. The Journal of biological chemistry. 1999;274(32):22739–22746. [PubMed: 10428857]
- 201.
- Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ. Molecular identification of the apical Ca2+ channel in 1, 25- dihydroxyvitamin D3-responsive epithelia. The Journal of biological chemistry. 1999;274(13):8375–8378. [PubMed: 10085067]
- 202.
- Peng JB, Chen XZ, Berger UV, Vassilev PM, Brown EM, Hediger MA. A rat kidney-specific calcium transporter in the distal nephron. The Journal of biological chemistry. 2000;275(36):28186–28194. [PubMed: 10875938]
- 203.
- Muller D, Hoenderop JG, Meij IC, van den Heuvel LP, Knoers NV, den Hollander AI, Eggert P, Garcia-Nieto V, Claverie-Martin F, Bindels RJ. Molecular cloning, tissue distribution, and chromosomal mapping of the human epithelial Ca2+ channel (ECAC1). Genomics. 2000;67(1):48–53. [PubMed: 10945469]
- 204.
- Peng JB, Brown EM, Hediger MA. Structural conservation of the genes encoding cat1, cat2, and related cation channels. Genomics. 2001;76(1-3):99–109. [PubMed: 11549322]
- 205.
- Song Y, Peng X, Porta A, Takanaga H, Peng JB, Hediger MA, Fleet JC, Christakos S. Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology. 2003;144(9):3885–3894. [PubMed: 12933662]
- 206.
- Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, Zhuang L, Freeman MR, Gouveia CH, Wu J, Luo H, Mauro T, Brown EM, Hediger MA. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22(2):274-285. [PMC free article: PMC4548943] [PubMed: 17129178]
- 207.
- Sampson HW, Matthews JL, Martin JH, Kunin AS. An electron microscopic localization of calcium in the small intestine of normal, rachitic, and vitamin-D-treated rats. Calcif Tissue Res. 1970;5(4):305–316. [PubMed: 5469610]
- 208.
- Schaefer HJ. Ultrastructure and ion distribution of the intestinal cell during experimental vitamin D deficiency rickets in rats. Virchows Archiv. 1973;359:111–123. [PubMed: 4196364]
- 209.
- Chandra S, Fullmer CS, Smith CA, Wasserman RH, Morrison GH. Ion microscopic imaging of calcium transport in the intestinal tissue of vitamin D-deficient and vitamin D-replete chickens: a 44Ca stable isotope study. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(15):5715–5719. [PMC free article: PMC54398] [PubMed: 2377608]
- 210.
- Bikle DD, Zolock DT, Morrissey RL, Herman RH. Independence of 1,25-dihydroxyvitamin D3-mediated calcium transport from de novo RNA and protein synthesis. The Journal of biological chemistry. 1978;253(2):484–488. [PubMed: 618881]
- 211.
- Bikle DD, Morrissey RL, Zolock DT. The mechanism of action of vitamin D in the intestine. Am J Clin Nutr. 1979;32(11):2322–2328. [PubMed: 158978]
- 212.
- Glenney JR Jr, Glenney P. Comparison of Ca++-regulated events in the intestinal brush border. J Cell Biol. 1985;100(3):754–763. [PMC free article: PMC2113509] [PubMed: 4038709]
- 213.
- Bikle DD, Gee E. Free, and not total, 1,25-dihydroxyvitamin D regulates 25- hydroxyvitamin D metabolism by keratinocytes. Endocrinology. 1989;124(2):649–654. [PubMed: 2463902]
- 214.
- Bikle DD, Munson S, Chafouleas J. Calmodulin may mediate 1,25-dihydroxyvitamin D-stimulated intestinal calcium transport. FEBS Lett. 1984;174(1):30–33. [PubMed: 6547914]
- 215.
- Howe CL, Keller TC 3rd, Mooseker MS, Wasserman RH. Analysis of cytoskeletal proteins and Ca2+-dependent regulation of structure in intestinal brush borders from rachitic chicks. Proc Natl Acad Sci U S A. 1982;79(4):1134–1138. [PMC free article: PMC345915] [PubMed: 6951164]
- 216.
- Bikle DD, Munson S. The villus gradient of brush border membrane calmodulin and the calcium-independent calmodulin-binding protein parallels that of calcium-accumulating ability. Endocrinology. 1986;118(2):727–732. [PubMed: 3753677]
- 217.
- Drenckhahn D, Dermietzel R. Organization of the actin filament cytoskeleton in the intestinal brush border: a quantitative and qualitative immunoelectron microscope study. J Cell Biol. 1988;107(3):1037–1048. [PMC free article: PMC2115304] [PubMed: 3417773]
- 218.
- Munson S, Wang Y, Chang W, Bikle DD. Myosin 1a Regulates Osteoblast Differentiation Independent of Intestinal Calcium Transport. J Endocr Soc. 2019;3(11):1993–2011. [PMC free article: PMC6789431] [PubMed: 31620669]
- 219.
- Wasserman RH, Taylor AN. Vitamin D-dependent calcium-binding protein. Response to some physiological and nutritional variables. The Journal of biological chemistry. 1968;243(14):3987–3993. [PubMed: 4298517]
- 220.
- Lee GS, Lee KY, Choi KC, Ryu YH, Paik SG, Oh GT, Jeung EB. Phenotype of a calbindin-D9k gene knockout is compensated for by the induction of other calcium transporter genes in a mouse model. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22(12):1968-1978. [PubMed: 17696760]
- 221.
- Ghijsen WE, De Jong MD, Van Os CH. ATP-dependent calcium transport and its correlation with Ca2+ -ATPase activity in basolateral plasma membranes of rat duodenum. Biochim Biophys Acta. 1982;689(2):327–336. [PubMed: 6214277]
- 222.
- Cai Q, Chandler JS, Wasserman RH, Kumar R, Penniston JT. Vitamin D and adaptation to dietary calcium and phosphate deficiencies increase intestinal plasma membrane calcium pump gene expression. Proc Natl Acad Sci U S A. 1993;90(4):1345–1349. [PMC free article: PMC45869] [PubMed: 7679502]
- 223.
- Wasserman RH, Chandler JS, Meyer SA, Smith CA, Brindak ME, Fullmer CS, Penniston JT, Kumar R. Intestinal calcium transport and calcium extrusion processes at the basolateral membrane. J Nutr. 1992;122(3) Suppl:662–671. [PubMed: 1311756]
- 224.
- Ryan ZC, Craig TA, Filoteo AG, Westendorf JJ, Cartwright EJ, Neyses L, Strehler EE, Kumar R. Deletion of the intestinal plasma membrane calcium pump, isoform 1, Atp2b1, in mice is associated with decreased bone mineral density and impaired responsiveness to 1, 25-dihydroxyvitamin D3. Biochem Biophys Res Commun. 2015;467(1):152–156. [PMC free article: PMC4772868] [PubMed: 26392310]
- 225.
- Liu C, Weng H, Chen L, Yang S, Wang H, Debnath G, Guo X, Wu L, Mohandas N, An X. Impaired intestinal calcium absorption in protein 4.1R-deficient mice due to altered expression of plasma membrane calcium ATPase 1b (PMCA1b). The Journal of biological chemistry. 2013;288(16):11407–11415. [PMC free article: PMC3630858] [PubMed: 23460639]
- 226.
- Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85(1):373–422. [PubMed: 15618484]
- 227.
- Charoenphandhu N, Krishnamra N. Prolactin is an important regulator of intestinal calcium transport. Can J Physiol Pharmacol. 2007;85(6):569–581. [PubMed: 17823618]
- 228.
- Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273(4):C1378–1385. [PubMed: 9357784]
- 229.
- Fujita H, Chiba H, Yokozaki H, Sakai N, Sugimoto K, Wada T, Kojima T, Yamashita T, Sawada N. Differential expression and subcellular localization of claudin-7, -8, -12, -13, and -15 along the mouse intestine. J Histochem Cytochem. 2006;54(8):933–944. [PubMed: 16651389]
- 230.
- Van Itallie CM, Fanning AS, Anderson JM. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol. 2003;285(6):F1078–1084. [PubMed: 13129853]
- 231.
- Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, Yamamoto Y, Wada T, Kojima T, Yokozaki H, Yamashita T, Kato S, Sawada N, Chiba H. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell. 2008;19(5):1912–1921. [PMC free article: PMC2366872] [PubMed: 18287530]
- 232.
- Charoenphandhu N, Nakkrasae LI, Kraidith K, Teerapornpuntakit J, Thongchote K, Thongon N, Krishnamra N. Two-step stimulation of intestinal Ca(2+) absorption during lactation by long-term prolactin exposure and suckling-induced prolactin surge. Am J Physiol Endocrinol Metab. 2009;297(3):E609–619. [PubMed: 19567804]
- 233.
- Harrison HE, Harrison HC. Intestinal transport of phosphate: Action of vitamin D, calcium, and potassium. American Journal of Physiology. 1961;201:1007–1012. [PubMed: 13904900]
- 234.
- Peterlik M, Wasserman RH. Regulation by vitamin D of intestinal phosphate absorption. Horm Metab Res. 1980;12(5):216–219. [PubMed: 6248445]
- 235.
- Xu H, Bai L, Collins JF, Ghishan FK. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics. 1999;62(2):281–284. [PubMed: 10610722]
- 236.
- Xu H, Bai L, Collins JF, Ghishan FK. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). American journal of physiology Cell physiology. 2002;282(3):C487–493. [PubMed: 11832333]
- 237.
- Fuchs R, Peterlik M. Vitamin D-induced transepithelial phosphate and calcium transport by chick jejunum. Effect of microfilamentous and microtubular inhibitors. FEBS Lett. 1979;100(2):357–359. [PubMed: 222609]
- 238.
- Narbaitz R, Stumpf WE, Sar M, Huang S, DeLuca HF. Autoradiographic localization of target cells for 1 alpha, 25- dihydroxyvitamin D3 in bones from fetal rats. Calcif Tissue Int. 1983;35(2):177–182. [PubMed: 6687827]
- 239.
- Boivin G, Mesguich P, Pike JW, Bouillon R, Meunier PJ, Haussler MR, Dubois PM, Morel G. Ultrastructural immunocytochemical localization of endogenous 1,25- dihydroxyvitamin D3 and its receptors in osteoblasts and osteocytes from neonatal mouse and rat calvaria. Bone Miner. 1987;3(2):125–136. [PubMed: 2850050]
- 240.
- Nakamichi Y, Udagawa N, Horibe K, Mizoguchi T, Yamamoto Y, Nakamura T, Hosoya A, Kato S, Suda T, Takahashi N. VDR in Osteoblast-Lineage Cells Primarily Mediates Vitamin D Treatment-Induced Increase in Bone Mass by Suppressing Bone Resorption. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2017;32(6):1297-1308. [PubMed: 28177161]
- 241.
- Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, Goltzman D. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. The Journal of biological chemistry. 2004;279(16):16754–16766. [PubMed: 14739296]
- 242.
- Chenu C, Valentin-Opran A, Chavassieux P, Saez S, Meunier PJ, Delmas PD. Insulin like growth factor I hormonal regulation by growth hormone and by 1,25(OH)2D3 and activity on human osteoblast-like cells in short- term cultures. Bone. 1990;11(2):81–86. [PubMed: 2357426]
- 243.
- Kurose H, Yamaoka K, Okada S, Nakajima S, Seino Y. 1,25-Dihydroxyvitamin D3 [1,25-(OH)2D3] increases insulin-like growth factor I (IGF-I) receptors in clonal osteoblastic cells. Study on interaction of IGF-I and 1,25-(OH)2D3. Endocrinology. 1990;126(4):2088–2094. [PubMed: 2156680]
- 244.
- Scharla SH, Strong DD, Mohan S, Baylink DJ, Linkhart TA. 1,25-Dihydroxyvitamin D3 differentially regulates the production of insulin-like growth factor I (IGF-I) and IGF-binding protein-4 in mouse osteoblasts. Endocrinology. 1991;129(6):3139–3146. [PubMed: 1720089]
- 245.
- Moriwake T, Tanaka H, Kanzaki S, Higuchi J, Seino Y. 1,25-Dihydroxyvitamin D3 stimulates the secretion of insulin-like growth factor binding protein 3 (IGFBP-3) by cultured human osteosarcoma cells. Endocrinology. 1992;130(2):1071–1073. [PubMed: 1370789]
- 246.
- Sato T, Ono T, Tuan RS. 1,25-Dihydroxy vitamin D3 stimulation of TGF-beta expression in chick embryonic calvarial bone. Differentiation. 1993;52(2):139–150. [PubMed: 8472884]
- 247.
- Wang DS, Yamazaki K, Nohtomi K, Shizume K, Ohsumi K, Shibuya M, Demura H, Sato K. Increase of vascular endothelial growth factor mRNA expression by 1,25- dihydroxyvitamin D3 in human osteoblast-like cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1996;11(4):472-479. [PubMed: 8992878]
- 248.
- Lacey DL, Grosso LE, Moser SA, Erdmann J, Tan HL, Pacifici R, Villareal DT. IL-1-induced murine osteoblast IL-6 production is mediated by the type 1 IL-1 receptor and is increased by 1,25 dihydroxyvitamin D3. The Journal of clinical investigation. 1993;91(4):1731–1742. [PMC free article: PMC288153] [PubMed: 8473513]
- 249.
- Lacey DL, Erdmann JM, Tan HL, Ohara J. Murine osteoblast interleukin 4 receptor expression: upregulation by 1,25 dihydroxyvitamin D3. Journal of cellular biochemistry. 1993;53(2):122–134. [PubMed: 8227185]
- 250.
- Nambi P, Wu HL, Lipshutz D, Prabhakar U. Identification and characterization of endothelin receptors on rat osteoblastic osteosarcoma cells: down-regulation by 1,25-dihydroxy- vitamin D3. Mol Pharmacol. 1995;47(2):266–271. [PubMed: 7870034]
- 251.
- Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10(12):e1004820. [PMC free article: PMC4256265] [PubMed: 25474590]
- 252.
- Johnson JA, Grande JP, Roche PC, Kumar R. Ontogeny of the 1,25-dihydroxyvitamin D3 receptor in fetal rat bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1996;11(1):56-61. [PubMed: 8770697]
- 253.
- Miller SC, Halloran BP, DeLuca HF, Lee WSS. Studies on the role of vitamin D in early skeletal development, mineralization, and growth in rats. Calcif Tissue Int. 1983;35:455–460. [PubMed: 6616315]
- 254.
- Kyeyune-Nyombi E, Lau KH, Baylink DJ, Strong DD. 1,25-Dihydroxyvitamin D3 stimulates both alkaline phosphatase gene transcription and mRNA stability in human bone cells. Arch Biochem Biophys. 1991;291(2):316–325. [PubMed: 1952946]
- 255.
- Irving JT, Wuthier RE. Histochemistry and biochemistry of calcification with special reference to the role of lipids. Clin Orthop. 1968;(56):237–260. [PubMed: 4172398]
- 256.
- Howell DS, Marquez JF, Pita JC. The nature of phospholipids in normal and rachitic costochondral plates. Arthritis Rheum. 1965;8(6):1039–1046. [PubMed: 5884814]
- 257.
- Dean DD, Boyan BD, Muniz OE, Howell DS, Schwartz Z. Vitamin D metabolites regulate matrix vesicle metalloproteinase content in a cell maturation-dependent manner. Calcif Tissue Int. 1996;59(2):109–116. [PubMed: 8687979]
- 258.
- Roughley PJ, Dickson IR. A comparison of proteoglycan from chick cartilage of different types and a study of the effect of vitamin D on proteoglycan structure. Connect Tissue Res. 1986;14(3):187–197. [PubMed: 3009087]
- 259.
- Plachot JJ, Du Bois MB, Halpern S, Cournot-Witmer G, Garabedian M, Balsan S. In vitro action of 1,25-dihydroxycholecalciferol and 24,25- dihydroxycholecalciferol on matrix organization and mineral distribution in rabbit growth plate. Metab Bone Dis Relat Res. 1982;4(2):135–142. [PubMed: 6983024]
- 260.
- Boyan BD, Schwartz Z, Carnes DL Jr, Ramirez V. The effects of vitamin D metabolites on the plasma and matrix vesicle membranes of growth and resting cartilage cells in vitro. Endocrinology. 1988;122(6):2851–2860. [PubMed: 2836176]
- 261.
- Schwartz Z, Boyan B. The effects of vitamin D metabolites on phospholipase A2 activity of growth zone and resting zone cartilage cells in vitro. Endocrinology. 1988;122(5):2191–2198. [PubMed: 3258818]
- 262.
- Swain LD, Schwartz Z, Caulfield K, Brooks BP, Boyan BD. Nongenomic regulation of chondrocyte membrane fluidity by 1,25-(OH)2D3 and 24,25-(OH)2D3 is dependent on cell maturation. Bone. 1993;14(4):609–617. [PubMed: 8274303]
- 263.
- Sylvia VL, Schwartz Z, Schuman L, Morgan RT, Mackey S, Gomez R, Boyan BD. Maturation-dependent regulation of protein kinase C activity by vitamin D3 metabolites in chondrocyte cultures. J Cell Physiol. 1993;157(2):271–278. [PubMed: 8227160]
- 264.
- Boyan BD, Chen J, Schwartz Z. Mechanism of Pdia3-dependent 1alpha,25-dihydroxy vitamin D3 signaling in musculoskeletal cells. Steroids. 2012;77(10):892–896. [PubMed: 22569272]
- 265.
- Owen TA, Aronow MS, Barone LM, Bettencourt B, Stein GS, Lian JB. Pleiotropic effects of vitamin D on osteoblast gene expression are related to the proliferative and differentiated state of the bone cell phenotype: dependency upon basal levels of gene expression, duration of exposure, and bone matrix competency in normal rat osteoblast cultures. Endocrinology. 1991;128(3):1496–1504. [PubMed: 1999168]
- 266.
- Lian J, Stewart C, Puchacz E, Mackowiak S, Shalhoub V, Collart D, Zambetti G, Stein G. Structure of the rat osteocalcin gene and regulation of vitamin D- dependent expression. Proc Natl Acad Sci U S A. 1989;86(4):1143–1147. [PMC free article: PMC286642] [PubMed: 2784002]
- 267.
- Manolagas SC, Burton DW, Deftos LJ. 1,25-Dihydroxyvitamin D3 stimulates the alkaline phosphatase activity of osteoblast-like cells. The Journal of biological chemistry. 1981;256(14):7115–7117. [PubMed: 6941964]
- 268.
- Rowe DW, Kream BE. Regulation of collagen synthesis in fetal rat calvaria by 1,25- dihydroxyvitamin D3. The Journal of biological chemistry. 1982;257(14):8009–8015. [PubMed: 6896329]
- 269.
- Prince CW, Butler WT. 1,25-Dihydroxyvitamin D3 regulates the biosynthesis of osteopontin, a bone-derived cell attachment protein, in clonal osteoblast-like osteosarcoma cells. Coll Relat Res. 1987;7(4):305–313. [PubMed: 3478171]
- 270.
- Demay MB, Gerardi JM, DeLuca HF, Kronenberg HM. DNA sequences in the rat osteocalcin gene that bind the 1,25- dihydroxyvitamin D3 receptor and confer responsiveness to 1,25- dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1990;87(1):369–373. [PMC free article: PMC53265] [PubMed: 2153298]
- 271.
- Kerner SA, Scott RA, Pike JW. Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(12):4455–4459. [PMC free article: PMC287288] [PubMed: 2786632]
- 272.
- Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25- dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87(24):9995–9999. [PMC free article: PMC55301] [PubMed: 2175918]
- 273.
- Zhang R, Ducy P, Karsenty G. 1,25-dihydroxyvitamin D3 inhibits Osteocalcin expression in mouse through an indirect mechanism. The Journal of biological chemistry. 1997;272(1):110–116. [PubMed: 8995235]
- 274.
- Wronski TJ, Halloran BP, Bikle DD, Globus RK, Morey-Holton ER. Chronic administration of 1,25-dihydroxyvitamin D3: increased bone but impaired mineralization. Endocrinology. 1986;119(6):2580–2585. [PubMed: 3780540]
- 275.
- Hock JM, Gunness-Hey M, Poser J, Olson H, Bell NH, Raisz LG. Stimulation of undermineralized matrix formation by 1,25 dihydroxyvitamin D3 in long bones of rats. Calcif Tissue Int. 1986;38(2):79–86. [PubMed: 3082498]
- 276.
- Suda T, Takahashi N, Abe E. Role of vitamin D in bone resorption. Journal of cellular biochemistry. 1992;49(1):53–58. [PubMed: 1644855]
- 277.
- Merke J, Klaus G, Hugel U, Waldherr R, Ritz E. No 1,25-dihydroxyvitamin D3 receptors on osteoclasts of calcium- deficient chicken despite demonstrable receptors on circulating monocytes. The Journal of clinical investigation. 1986;77(1):312–314. [PMC free article: PMC423341] [PubMed: 3003152]
- 278.
- Mee AP, Hoyland JA, Braidman IP, Freemont AJ, Davies M, Mawer EB. Demonstration of vitamin D receptor transcripts in actively resorbing osteoclasts in bone sections. Bone. 1996;18(4):295–299. [PubMed: 8726384]
- 279.
- Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption--a hypothesis. Calcif Tissue Int. 1981;33(4):349–351. [PubMed: 6271355]
- 280.
- Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–357. [PubMed: 10368775]
- 281.
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597–3602. [PMC free article: PMC19881] [PubMed: 9520411]
- 282.
- Takeda S, Yoshizawa T, Nagai Y, Yamato H, Fukumoto S, Sekine K, Kato S, Matsumoto T, Fujita T. Stimulation of osteoclast formation by 1,25-dihydroxyvitamin D requires its binding to vitamin D receptor (VDR) in osteoblastic cells: studies using VDR knockout mice. Endocrinology. 1999;140(2):1005–1008. [PubMed: 9927335]
- 283.
- Friedman PA, Gesek FA. Cellular calcium transport in renal epithelia: measurement, mechanisms, and regulation. Physiol Rev. 1995;75(3):429–471. [PubMed: 7624390]
- 284.
- Winaver J, Sylk DB, Robertson JS, Chen TC, Puschett JB. Micropuncture study of the acute renal tubular transport effects of 25-hydroxyvitamin D3 in the dog. Mineral and electrolyte metabolism. 1980;4:178–188.
- 285.
- Tenenhouse HS. Cellular and molecular mechanisms of renal phosphate transport. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1997;12(2):159-164. [PubMed: 9041046]
- 286.
- Puschett JB, Beck WS Jr, Jelonek A, Fernandez PC. Study of the renal tubular interactions of thyrocalcitonin, cyclic adenosine 3',5'-monophosphate, 25-hydroxycholecalciferol, and calcium ion. The Journal of clinical investigation. 1974;53(3):756–767. [PMC free article: PMC333056] [PubMed: 4359939]
- 287.
- Puschett JB, Fernandez PC, Boyle IT, Gray RW, Omdahl JL, DeLuca HF. The acute renal tubular effects of 1,25-dihydroxycholecalciferol. Proc Soc Exp Biol Med. 1972;141(1):379–384. [PubMed: 5082314]
- 288.
- Puschett JB, Moranz J, Kurnick WS. Evidence for a direct action of cholecalciferol and 25- hydroxycholecalciferol on the renal transport of phosphate, sodium, and calcium. The Journal of clinical investigation. 1972;51(2):373–385. [PMC free article: PMC302136] [PubMed: 4333022]
- 289.
- Popovtzer MM, Robinette JB, DeLuca HF, Holick MF. The acute effect of 25-hydroxycholecalciferol on renal handling of phosphorus. Evidence for a parathyroid hormone-dependent mechanism. The Journal of clinical investigation. 1974;53(3):913–921. [PMC free article: PMC333074] [PubMed: 4812447]
- 290.
- Yamamoto M, Kawanobe Y, Takahashi H, Shimazawa E, Kimura S, Ogata E. Vitamin D deficiency and renal calcium transport in the rat. The Journal of clinical investigation. 1984;74(2):507–513. [PMC free article: PMC370503] [PubMed: 6086715]
- 291.
- Kumar R, Schaefer J, Grande JP, Roche PC. Immunolocalization of calcitriol receptor, 24-hydroxylase cytochrome P- 450, and calbindin D28k in human kidney. Am J Physiol. 1994;266(3 Pt 2):F477–485. [PubMed: 8160797]
- 292.
- Borke JL, Minami J, Verma AK, Penniston JT, Kumar R. Co-localization of erythrocyte Ca++-Mg++ ATPase and vitamin D-dependent 28-kDa-calcium binding protein. Kidney Int. 1988;34(2):262–267. [PubMed: 2460662]
- 293.
- Christakos S, Brunette MG, Norman AW. Localization of immunoreactive vitamin D-dependent calcium binding protein in chick nephron. Endocrinology. 1981;109(1):322–324. [PubMed: 7238412]
- 294.
- Roth J, Thorens B, Hunziker W, Norman AW, Orci L. Vitamin D--dependent calcium binding protein: immunocytochemical localization in chick kidney. Science. 1981;214(4517):197–200. [PubMed: 7025212]
- 295.
- Bouhtiauy I, Lajeunesse D, Christakos S, Brunette MG. Two vitamin D3-dependent calcium binding proteins increase calcium reabsorption by different mechanisms. I. Effect of CaBP 28K. Kidney Int. 1994;45(2):461–468. [PubMed: 8164434]
- 296.
- Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol. 2013;75:535–550. [PubMed: 23398154]
- 297.
- Silver J, Russell J, Sherwood LM. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci U S A. 1985;82(12):4270–4273. [PMC free article: PMC397979] [PubMed: 3858880]
- 298.
- Cantley LK, Russell J, Lettieri D, Sherwood LM. 1,25-Dihydroxyvitamin D3 suppresses parathyroid hormone secretion from bovine parathyroid cells in tissue culture. Endocrinology. 1985;117(5):2114–2119. [PubMed: 3840080]
- 299.
- Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM. Sequences in the human parathyroid hormone gene that bind the 1,25- dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1992;89(17):8097–8101. [PMC free article: PMC49863] [PubMed: 1325645]
- 300.
- Russell J, Ashok S, Koszewski NJ. Vitamin D receptor interactions with the rat parathyroid hormone gene: synergistic effects between two negative vitamin D response elements. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1999;14(11):1828-1837. [PubMed: 10571682]
- 301.
- Nishishita T, Okazaki T, Ishikawa T, Igarashi T, Hata K, Ogata E, Fujita T. A negative vitamin D response DNA element in the human parathyroid hormone-related peptide gene binds to vitamin D receptor along with Ku antigen to mediate negative gene regulation by vitamin D. The Journal of biological chemistry. 1998;273(18):10901–10907. [PubMed: 9556566]
- 302.
- Hawa NS, O'Riordan JL, Farrow SM. Functional analysis of vitamin D response elements in the parathyroid hormone gene and a comparison with the osteocalcin gene. Biochem Biophys Res Commun. 1996;228(2):352–357. [PubMed: 8920918]
- 303.
- Mackey SL, Heymont JL, Kronenberg HM, Demay MB. Vitamin D receptor binding to the negative human parathyroid hormone vitamin D response element does not require the retinoid x receptor. Mol Endocrinol. 1996;10(3):298–305. [PubMed: 8833658]
- 304.
- Canaff L, Hendy GN. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. The Journal of biological chemistry. 2002;277(33):30337–30350. [PubMed: 12036954]
- 305.
- Naveh-Many T, Marx R, Keshet E, Pike JW, Silver J. Regulation of 1,25-dihydroxyvitamin D3 receptor gene expression by 1,25- dihydroxyvitamin D3 in the parathyroid in vivo. The Journal of clinical investigation. 1990;86(6):1968–1975. [PMC free article: PMC329833] [PubMed: 2174913]
- 306.
- Russell J, Bar A, Sherwood LM, Hurwitz S. Interaction between calcium and 1,25-dihydroxyvitamin D3 in the regulation of preproparathyroid hormone and vitamin D receptor messenger ribonucleic acid in avian parathyroids. Endocrinology. 1993;132(6):2639–2644. [PubMed: 8389284]
- 307.
- Dedhar S, Rennie PS, Shago M, Hagesteijn CY, Yang H, Filmus J, Hawley RG, Bruchovsky N, Cheng H, Matusik RJ, et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nature. 1994;367(6462):480–483. [PubMed: 8107809]
- 308.
- Wheeler DG, Horsford J, Michalak M, White JH, Hendy GN. Calreticulin inhibits vitamin D3 signal transduction. Nucleic Acids Res. 1995;23(16):3268–3274. [PMC free article: PMC307187] [PubMed: 7667104]
- 309.
- Sela A, Silver J, Naveh-Many T. Chronic hypocalcemia increases PTH mRNA despite high 1,25(OH)2D levels:Roles of calretivulin and the vitamin D receptor. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1996;11: (abstract).
- 310.
- Ritter CS, Haughey BH, Armbrecht HJ, Brown AJ. Distribution and regulation of the 25-hydroxyvitamin D3 1alpha-hydroxylase in human parathyroid glands. J Steroid Biochem Mol Biol. 2012;130(1-2):73–80. [PubMed: 22326730]
- 311.
- Kadowaki S, Norman AW. Demonstration that the vitamin D metabolite 1,25(OH)2-vitamin D3 and not 24R,25(OH)2-vitamin D3 is essential for normal insulin secretion in the perfused rat pancreas. Diabetes. 1985;34(4):315–320. [PubMed: 2982684]
- 312.
- Lee S, Clark SA, Gill RK, Christakos S. 1,25-Dihydroxyvitamin D3 and pancreatic beta-cell function: vitamin D receptors, gene expression, and insulin secretion. Endocrinology. 1994;134(4):1602–1610. [PubMed: 8137721]
- 313.
- Clark SA, Stumpf WE, Sar M, DeLuca HF, Tanaka Y. Target cells for 1,25 dihydroxyvitamin D3 in the pancreas. Cell and tissue research. 1980;209(3):515–520. [PubMed: 6996826]
- 314.
- Morrissey RL, Bucci TJ, Richard B, Empson N, Lufkin EG. Calcium-binding protein: its cellular localization in jejunum, kidney and pancreas. Proc Soc Exp Biol Med. 1975;149(1):56–60. [PubMed: 806919]
- 315.
- Bland R, Markovic D, Hills CE, Hughes SV, Chan SL, Squires PE, Hewison M. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in pancreatic islets. The Journal of steroid biochemistry and molecular biology. 2004;89-90(1-5):121–125. [PubMed: 15225758]
- 316.
- Sooy K, Schermerhorn T, Noda M, Surana M, Rhoten WB, Meyer M, Fleischer N, Sharp GW, Christakos S. Calbindin-D(28k) controls [Ca(2+)](i) and insulin release. Evidence obtained from calbindin-d(28k) knockout mice and beta cell lines. The Journal of biological chemistry. 1999;274(48):34343–34349. [PubMed: 10567411]
- 317.
- Rabinovitch A, Suarez-Pinzon WL, Sooy K, Strynadka K, Christakos S. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology. 2001;142(8):3649–3655. [PubMed: 11459814]
- 318.
- Pittas AG, Lau J, Hu FB, Dawson-Hughes B. The role of vitamin D and calcium in type 2 diabetes. A systematic review and meta-analysis. The Journal of clinical endocrinology and metabolism. 2007;92(6):2017–2029. [PMC free article: PMC2085234] [PubMed: 17389701]
- 319.
- Pittas AG, Jorde R, Kawahara T, Dawson-Hughes B, Vitamin D. Supplementation for Prevention of Type 2 Diabetes Mellitus: To D or Not to D? The Journal of clinical endocrinology and metabolism. 2020;105(12) [PMC free article: PMC7571449] [PubMed: 32844212]
- 320.
- Pittas A, Dawson-Hughes B, Staten M., Vitamin D. Supplementation and Prevention of Type 2 Diabetes. N Engl J Med. 2019;381(18):1785–1786. Reply. [PubMed: 31665590]
- 321.
- Harris SS, Pittas AG, Palermo NJ. A randomized, placebo-controlled trial of vitamin D supplementation to improve glycaemia in overweight and obese African Americans. Diabetes Obes Metab. 2012;14(9):789–794. [PubMed: 22486948]
- 322.
- Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. American journal of physiology Gastrointestinal and liver physiology. 2005;289(6):G1036–1042. [PubMed: 16020653]
- 323.
- Fukumoto S, Yamashita T. FGF23 is a hormone-regulating phosphate metabolism--unique biological characteristics of FGF23. Bone. 2007;40(5):1190–1195. [PubMed: 17276744]
- 324.
- Eisman JA, Martin TJ, MacIntyre I, Moseley JM. 1,25-dihydroxyvitamin-D-receptor in breast cancer cells. Lancet. 1979;2(8156-8157):1335–1336. [PubMed: 92676]
- 325.
- Fleet JC, DeSmet M, Johnson R, Li Y. Vitamin D and cancer: a review of molecular mechanisms. Biochem J. 2012;441(1):61–76. [PMC free article: PMC4572477] [PubMed: 22168439]
- 326.
- Ingraham BA, Bragdon B, Nohe A. Molecular basis of the potential of vitamin D to prevent cancer. Current medical research and opinion. 2008;24(1):139–149. [PubMed: 18034918]
- 327.
- Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. The Journal of biological chemistry. 2003;278(48):48137–48145. [PubMed: 12972427]
- 328.
- Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, Zinser G, Valrance M, Aranda A, Moras D, Norman A, Welsh J, Byers SW. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Molecular cell. 2006;21(6):799–809. [PubMed: 16543149]
- 329.
- Dixon KM, Deo SS, Wong G, Slater M, Norman AW, Bishop JE, Posner GH, Ishizuka S, Halliday GM, Reeve VE, Mason RS. Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. The Journal of steroid biochemistry and molecular biology. 2005;97(1-2):137–143. [PubMed: 16039116]
- 330.
- Demetriou SK, Ona-Vu K, Teichert AE, Cleaver JE, Bikle DD, Oh DH. Vitamin D receptor mediates DNA repair and is UV inducible in intact epidermis but not in cultured keratinocytes. The Journal of investigative dermatology. 2012;132(8):2097–2100. [PMC free article: PMC3396713] [PubMed: 22495177]
- 331.
- De Haes P, Garmyn M, Degreef H, Vantieghem K, Bouillon R, Segaert S. 1,25-Dihydroxyvitamin D3 inhibits ultraviolet B-induced apoptosis, Jun kinase activation, and interleukin-6 production in primary human keratinocytes. J Cell Biochem. 2003;89(4):663–673. [PubMed: 12858333]
- 332.
- Gupta R, Dixon KM, Deo SS, Holliday CJ, Slater M, Halliday GM, Reeve VE, Mason RS. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. The Journal of investigative dermatology. 2007;127(3):707–715. [PubMed: 17170736]
- 333.
- Garland C, Shekelle RB, Barrett-Connor E, Criqui MH, Rossof AH, Paul O. Dietary vitamin D and calcium and risk of colorectal cancer: a 19-year prospective study in men. Lancet. 1985;1(8424):307–309. [PubMed: 2857364]
- 334.
- Bostick RM, Potter JD, Sellers TA, McKenzie DR, Kushi LH, Folsom AR. Relation of calcium, vitamin D, and dairy food intake to incidence of colon cancer among older women. The Iowa Women's Health Study. Am J Epidemiol. 1993;137(12):1302–1317. [PubMed: 8333412]
- 335.
- Kearney J, Giovannucci E, Rimm EB, Ascherio A, Stampfer MJ, Colditz GA, Wing A, Kampman E, Willett WC. Calcium, vitamin D, and dairy foods and the occurrence of colon cancer in men. Am J Epidemiol. 1996;143(9):907–917. [PubMed: 8610704]
- 336.
- Garland FC, Garland CF, Gorham ED, Young JF. Geographic variation in breast cancer mortality in the United States: a hypothesis involving exposure to solar radiation. Prev Med. 1990;19(6):614–622. [PubMed: 2263572]
- 337.
- Hanchette CL, Schwartz GG. Geographic patterns of prostate cancer mortality. Evidence for a protective effect of ultraviolet radiation. Cancer. 1992;70(12):2861–2869. [PubMed: 1451068]
- 338.
- Boscoe FP, Schymura MJ. Solar ultraviolet-B exposure and cancer incidence and mortality in the United States, 1993-2002. BMC Cancer. 2006;6:264. [PMC free article: PMC1665523] [PubMed: 17096841]
- 339.
- Diercke K, Kohl A, Lux CJ, Erber R. Strain-dependent up-regulation of ephrin-B2 protein in periodontal ligament fibroblasts contributes to osteogenesis during tooth movement. The Journal of biological chemistry. 2011;286(43):37651–37664. [PMC free article: PMC3199509] [PubMed: 21880727]
- 340.
- Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. The American journal of clinical nutrition. 2007;85(6):1586–1591. [PubMed: 17556697]
- 341.
- Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Gordon D, Copeland T, D'Agostino D, Friedenberg G, Ridge C, Bubes V, Giovannucci EL, Willett WC, Buring JE, Group VR, Vitamin D. Supplements and Prevention of Cancer and Cardiovascular Disease. N Engl J Med. 2019;380(1):33–44. [PMC free article: PMC6425757] [PubMed: 30415629]
- 342.
- Gross C, Stamey T, Hancock S, Feldman D. Treatment of early recurrent prostate cancer with 1,25-dihydroxyvitamin D3 (calcitriol). J Urol. 1998;159(6):2035–2039. [PubMed: 9598513]
- 343.
- Beer TM, Ryan CW, Venner PM, Petrylak DP, Chatta GS, Ruether JD, Redfern CH, Fehrenbacher L, Saleh MN, Waterhouse DM, Carducci MA, Vicario D, Dreicer R, Higano CS, Ahmann FR, Chi KN, Henner WD, Arroyo A, Clow FW. Double-blinded randomized study of high-dose calcitriol plus docetaxel compared with placebo plus docetaxel in androgen-independent prostate cancer: a report from the ASCENT Investigators. J Clin Oncol. 2007;25(6):669–674. [PubMed: 17308271]
- 344.
- Barnett CM, Beer TM. Prostate cancer and vitamin D: what does the evidence really suggest? Urol Clin North Am. 2011;38(3):333–342. [PubMed: 21798396]
- 345.
- Lehmann B, Tiebel O, Meurer M. Expression of vitamin D3 25-hydroxylase (CYP27) mRNA after induction by vitamin D3 or UVB radiation in keratinocytes of human skin equivalents-- a preliminary study. Arch Dermatol Res. 1999;291(9):507–510. [PubMed: 10541881]
- 346.
- Bikle DD, Nemanic MK, Gee E, Elias P. 1,25-Dihydroxyvitamin D3 production by human keratinocytes. Kinetics and regulation. The Journal of clinical investigation. 1986;78(2):557–566. [PMC free article: PMC423594] [PubMed: 2426308]
- 347.
- Bikle DD, Pillai S, Gee E, Hincenbergs M. Tumor necrosis factor-alpha regulation of 1,25-dihydroxyvitamin D production by human keratinocytes. Endocrinology. 1991;129(1):33–38. [PubMed: 1675987]
- 348.
- Bikle DD, Pillai S, Gee E, Hincenbergs M. Regulation of 1,25-dihydroxyvitamin D production in human keratinocytes by interferon-gamma. Endocrinology. 1989;124(2):655–660. [PubMed: 2463903]
- 349.
- Pillai S, Bikle DD, Elias PM. 1,25-Dihydroxyvitamin D production and receptor binding in human keratinocytes varies with differentiation. The Journal of biological chemistry. 1988;263(11):5390–5395. [PubMed: 2451669]
- 350.
- Bikle DD. Vitamin D and the skin: Physiology and pathophysiology. Reviews in endocrine & metabolic disorders. 2012;13(1):3–19. [PMC free article: PMC3687803] [PubMed: 21845365]
- 351.
- Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980;19(1):245–254. [PubMed: 6153576]
- 352.
- Hennings H, Holbrook KA. Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Experimental cell research. 1983;143(1):127–142. [PubMed: 6186504]
- 353.
- Hennings H, Holbrook KA, Yuspa SH. Factors influencing calcium-induced terminal differentiation in cultured mouse epidermal cells. Journal of cellular physiology. 1983;116(3):265–281. [PubMed: 6885930]
- 354.
- Praeger FC, Stanulis-Praeger BM, Gilchrest BA. Use of strontium to separate calcium-dependent pathways for proliferation and differentiation in human keratinocytes. J Cell Physiol. 1987;132(1):81–89. [PubMed: 2439523]
- 355.
- Pillai S, Bikle DD, Mancianti ML, Cline P, Hincenbergs M. Calcium regulation of growth and differentiation of normal human keratinocytes: modulation of differentiation competence by stages of growth and extracellular calcium. Journal of Cellular Physiology. 1990;143(2):294–302. [PubMed: 1970572]
- 356.
- Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. The Journal of cell biology. 1989;109(3):1207–1217. [PMC free article: PMC2115750] [PubMed: 2475508]
- 357.
- Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert review of endocrinology & metabolism. 2012;7(4):461–472. [PMC free article: PMC3491811] [PubMed: 23144648]
- 358.
- Sheu HM, Kitajima Y, Yaoita H. Involvement of protein kinase C in translocation of desmoplakins from cytosol to plasma membrane during desmosome formation in human squamous cell carcinoma cells grown in low to normal calcium concentration. Exp Cell Res. 1989;185(1):176–190. [PubMed: 2509226]
- 359.
- Denning MF, Dlugosz AA, Williams EK, Szallasi Z, Blumberg PM, Yuspa SH. Specific protein kinase C isozymes mediate the induction of keratinocyte differentiation markers by calcium. Cell Growth Differ. 1995;6(2):149–157. [PubMed: 7756173]
- 360.
- Filvaroff E, Calautti E, McCormick F, Dotto GP. Specific changes of Ras GTPase-activating protein (GAP) and a GAP- associated p62 protein during calcium-induced keratinocyte differentiation. Mol Cell Biol. 1992;12(12):5319–5328. [PMC free article: PMC360469] [PubMed: 1448067]
- 361.
- Filvaroff E, Calautti E, Reiss M, Dotto GP. Functional evidence for an extracellular calcium receptor mechanism triggering tyrosine kinase activation associated with mouse keratinocyte differentiation. The Journal of biological chemistry. 1994;269(34):21735–21740. [PubMed: 8063817]
- 362.
- Rice RH, Green H. Presence in human epidermal cells of a soluble protein precursor of the cross-linked envelope: activation of the cross-linking by calcium ions. Cell. 1979;18(3):681–694. [PubMed: 42494]
- 363.
- Hohl D, Lichti U, Breitkreutz D, Steinert PM, Roop DR. Transcription of the human loricrin gene in vitro is induced by calcium and cell density and suppressed by retinoic acid. The Journal of investigative dermatology. 1991;96(4):414–418. [PubMed: 2007780]
- 364.
- Simon M, Green H. Participation of membrane-associated proteins in the formation of the cross-linked envelope of the keratinocyte. Cell. 1984;36(4):827–834. [PubMed: 6200236]
- 365.
- Hohl D. Cornified cell envelope. Dermatologica. 1990;180(4):201–211. [PubMed: 2192924]
- 366.
- Thacher SM, Rice RH. Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell. 1985;40(3):685–695. [PubMed: 2578891]
- 367.
- Hennings H, Steinert P, Buxman MM. Calcium induction of transglutaminase and the formation of epsilon(gamma-glutamyl) lysine cross-links in cultured mouse epidermal cells. Biochemical and biophysical research communications. 1981;102(2):739–745. [PubMed: 6118152]
- 368.
- Hennings H, Kruszewski FH, Yuspa SH, Tucker RW. Intracellular calcium alterations in response to increased external calcium in normal and neoplastic keratinocytes. Carcinogenesis. 1989;10(4):777–780. [PubMed: 2702726]
- 369.
- Gibson DF, Ratnam AV, Bikle DD. Evidence for separate control mechanisms at the message, protein, and enzyme activation levels for transglutaminase during calcium-induced differentiation of normal and transformed human keratinocytes. Journal of Investigative Dermatology. 1996;106(1):154–161. [PubMed: 8592067]
- 370.
- Su MJ, Bikle DD, Mancianti ML, Pillai S. 1,25-Dihydroxyvitamin D3 potentiates the keratinocyte response to calcium. The Journal of biological chemistry. 1994;269(20):14723–14729. [PubMed: 7910167]
- 371.
- Menon GK, Grayson S, Elias PM. Ionic calcium reservoirs in mammalian epidermis: ultrastructural localization by ion-capture cytochemistry. Journal of Investigative Dermatology. 1985;84(6):508–512. [PubMed: 3998499]
- 372.
- Mauro T, Bench G, Sidderas-Haddad E, Feingold K, Elias P, Cullander C. Acute barrier perturbation abolishes the Ca2+ and K+ gradients in murine epidermis: quantitative measurement using PIXE. Journal of Investigative Dermatology. 1998;111(6):1198–1201. [PubMed: 9856840]
- 373.
- Lee SH, Elias PM, Feingold KR, Mauro T. A role for ions in barrier recovery after acute perturbation. The Journal of investigative dermatology. 1994;102(6):976–979. [PubMed: 8006464]
- 374.
- Menon GK, Price LF, Bommannan B, Elias PM, Feingold KR. Selective obliteration of the epidermal calcium gradient leads to enhanced lamellar body secretion. The Journal of investigative dermatology. 1994;102(5):789–795. [PubMed: 8176264]
- 375.
- Lee SH, Elias PM, Proksch E, Menon GK, Mao-Quiang M, Feingold KR. Calcium and potassium are important regulators of barrier homeostasis in murine epidermis. The Journal of clinical investigation. 1992;89(2):530–538. [PMC free article: PMC442884] [PubMed: 1737844]
- 376.
- Menon GK, Elias PM, Lee SH, Feingold KR. Localization of calcium in murine epidermis following disruption and repair of the permeability barrier. Cell and Tissue Research. 1992;270(3):503–512. [PubMed: 1486603]
- 377.
- Bikle DD, Ratnam A, Mauro T, Harris J, Pillai S. Changes in calcium responsiveness and handling during keratinocyte differentiation. Potential role of the calcium receptor. J Clin Invest. 1996;97(4):1085–1093. [PMC free article: PMC507156] [PubMed: 8613532]
- 378.
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca-2+-sensing receptor from bovine parathyroid. Nature. 1993;366(6455):575–580. [PubMed: 8255296]
- 379.
- Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC, Nemeth EF, Fuller F. Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. Journal of Biological Chemistry. 1995;270(21):12919–12925. [PubMed: 7759551]
- 380.
- Tu CL, Chang W, Bikle DD. The extracellular calcium-sensing receptor Is Required for calcium- induced differentiation in human keratinocytes. The Journal of biological chemistry. 2001;276(44):41079–41085. [PubMed: 11500521]
- 381.
- Tu C, Chang W, Xie Z, Bikle D. Inactivation of the Calcium Sensing Receptor Inhibits E-cadherin-mediated Cell-Cell Adhesion and Calcium-induced Differentiation in Human Epidermal Keratinocytes. J Biol Chem. 2008;283:3519–3528. [PubMed: 18065418]
- 382.
- Tu CL, Crumrine DA, Man MQ, Chang W, Elalieh H, You M, Elias PM, Bikle DD. Ablation of the calcium-sensing receptor in keratinocytes impairs epidermal differentiation and barrier function. The Journal of investigative dermatology. 2012;132(10):2350–2359. [PMC free article: PMC3434298] [PubMed: 22622426]
- 383.
- Oda Y, Tu CL, Pillai S, Bikle DD. The calcium sensing receptor and its alternatively spliced form in keratinocyte differentiation. Journal of Biological Chemistry. 1998;273(36):23344–23352. [PubMed: 9722568]
- 384.
- Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal. 2008;1(35):ra1. [PMC free article: PMC3538864] [PubMed: 18765830]
- 385.
- Jaken S, Yuspa SH. Early signals for keratinocyte differentiation: role of Ca2+-mediated inositol lipid metabolism in normal and neoplastic epidermal cells. Carcinogenesis. 1988;9(6):1033–1038. [PubMed: 2453303]
- 386.
- Punnonen K, Denning M, Lee E, Li L, Rhee SG, Yuspa SH. Keratinocyte differentiation is associated with changes in the expression and regulation of phospholipase C isoenzymes. The Journal of investigative dermatology. 1993;101(5):719–726. [PubMed: 8228334]
- 387.
- Xie Z, Bikle DD. Phospholipase C-gamma1 is required for calcium-induced keratinocyte differentiation. The Journal of biological chemistry. 1999;274(29):20421–20424. [PubMed: 10400667]
- 388.
- Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. The Journal of biological chemistry. 2007;282(12):8695–8703. [PubMed: 17242406]
- 389.
- Xie Z, Singleton PA, Bourguignon LY, Bikle DD. Calcium-induced human keratinocyte differentiation requires src- and fyn-mediated phosphatidylinositol 3-kinase-dependent activation of phospholipase C-gamma1. Molecular biology of the cell. 2005;16(7):3236–3246. [PMC free article: PMC1165407] [PubMed: 15872086]
- 390.
- Yuspa SH, Ben T, Hennings H, Lichti U. Divergent responses in epidermal basal cells exposed to the tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1982;42(6):2344–2349. [PubMed: 6122503]
- 391.
- Dlugosz AA, Yuspa SH. Protein kinase C regulates keratinocyte transglutaminase (TG-K) gene expression in cultured primary mouse epidermal keratinocytes induced to terminally differentiate by calcium. Journal of Investigative Dermatology. 1994;102(4):409–414. [PubMed: 7908680]
- 392.
- Lu B, Rothnagel JA, Longley MA, Tsai SY, Roop DR. Differentiation-specific expression of human keratin 1 is mediated by a composite AP-1/steroid hormone element. The Journal of biological chemistry. 1994;269(10):7443–7449. [PubMed: 7510286]
- 393.
- Ng DC, Shafaee S, Lee D, Bikle DD. Requirement of an AP-1 site in the calcium response region of the involucrin promoter. The Journal of biological chemistry. 2000;275(31):24080–24088. [PubMed: 10816578]
- 394.
- Bikle DD, Ng D, Oda Y, Hanley K, Feingold K, Xie Z. The vitamin D response element of the involucrin gene mediates its regulation by 1,25-dihydroxyvitamin D3. The Journal of investigative dermatology. 2002;119(5):1109–1113. [PubMed: 12445200]
- 395.
- Hosomi J, Hosoi J, Abe E, Suda T, Kuroki T. Regulation of terminal differentiation of cultured mouse epidermal cells by 1 alpha,25-dihydroxyvitamin D3. Endocrinology. 1983;113(6):1950–1957. [PubMed: 6196178]
- 396.
- Stumpf WE, Sar M, Reid FA, Tanaka Y, DeLuca HF. Target cells for 1,25-dihydroxyvitamin D3 in intestinal tract, stomach, kidney, skin, pituitary, and parathyroid. Science. 1979;206(4423):1188–1190. [PubMed: 505004]
- 397.
- Smith EL, Walworth NC, Holick MF. Effect of 1 alpha,25-dihydroxyvitamin D3 on the morphologic and biochemical differentiation of cultured human epidermal keratinocytes grown in serum-free conditions. The Journal of investigative dermatology. 1986;86(6):709–714. [PubMed: 2423618]
- 398.
- Pillai S, Bikle DD. Role of intracellular-free calcium in the cornified envelope formation of keratinocytes: differences in the mode of action of extracellular calcium and 1,25 dihydroxyvitamin D3. Journal of cellular physiology. 1991;146(1):94–100. [PubMed: 1990023]
- 399.
- McLane JA, Katz M, Abdelkader N. Effect of 1,25-dihydroxyvitamin D3 on human keratinocytes grown under different culture conditions. In Vitro Cell Dev Biol. 1990;26(4):379–387. [PubMed: 2345124]
- 400.
- McLaughlin JA, Cantley LC, Holick MF. 1,25(OH)2D3 increased calcium and phosphatidylinositol metabolism in differentiating cultured human keratinocytes. J Nutr Biochem. 1990;1:81–87. [PubMed: 15539189]
- 401.
- Ratnam AV, Bikle DD, Cho JK. 1,25 dihydroxyvitamin D3 enhances the calcium response of keratinocytes. Journal of Cellular Physiology. 1999;178(2):188–196. [PubMed: 10048583]
- 402.
- Pillai S, Bikle DD, Su MJ, Ratnam A, Abe J. 1,25-Dihydroxyvitamin D3 upregulates the phosphatidylinositol signaling pathway in human keratinocytes by increasing phospholipase C levels. The Journal of clinical investigation. 1995;96(1):602–609. [PMC free article: PMC185235] [PubMed: 7615834]
- 403.
- Xie Z, Bikle DD. Inhibition of 1,25-dihydroxyvitamin-D-induced keratinocyte differentiation by blocking the expression of phospholipase C-gamma1. The Journal of investigative dermatology. 2001;117(5):1250–1254. [PubMed: 11710940]
- 404.
- Vandenberghe M, Raphael M, Lehen'kyi V, Gordienko D, Hastie R, Oddos T, Rao A, Hogan PG, Skryma R, Prevarskaya N. ORAI1 calcium channel orchestrates skin homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):E4839–4848. [PMC free article: PMC3864283] [PubMed: 24277812]
- 405.
- Tu CL, Chang W, Bikle DD. The calcium-sensing receptor-dependent regulation of cell-cell adhesion and keratinocyte differentiation requires Rho and filamin A. The Journal of investigative dermatology. 2011;131(5):1119–1128. [PMC free article: PMC3078217] [PubMed: 21209619]
- 406.
- Tu CL, Chang W, Bikle DD. Phospholipase cgamma1 is required for activation of store-operated channels in human keratinocytes. The Journal of investigative dermatology. 2005;124(1):187–197. [PubMed: 15654973]
- 407.
- Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocrine reviews. 1999;20(2):156–188. [PubMed: 10204116]
- 408.
- Bikle DD, Chang S, Crumrine D, Elalieh H, Man MQ, Choi EH, Dardenne O, Xie Z, Arnaud RS, Feingold K, Elias PM. 25 Hydroxyvitamin D 1 alpha-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. The Journal of investigative dermatology. 2004;122(4):984–992. [PubMed: 15102089]
- 409.
- Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(22):9428–9433. [PMC free article: PMC1890511] [PubMed: 17517646]
- 410.
- Palmer HG, Martinez D, Carmeliet G, Watt FM. The vitamin D receptor is required for mouse hair cycle progression but not for maintenance of the epidermal stem cell compartment. The Journal of investigative dermatology. 2008;128(8):2113–2117. [PubMed: 18337835]
- 411.
- Plikus MV, Gay DL, Treffeisen E, Wang A, Supapannachart RJ, Cotsarelis G. Epithelial stem cells and implications for wound repair. Seminars in cell & developmental biology. 2012;23(9):946–953. [PMC free article: PMC3518754] [PubMed: 23085626]
- 412.
- Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, Simons BD, Blanpain C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489(7415):257–262. [PubMed: 22940863]
- 413.
- Tian XQ, Chen TC, Holick MF. 1,25-dihydroxyvitamin D3: a novel agent for enhancing wound healing. Journal of cellular biochemistry. 1995;59(1):53–56. [PubMed: 8530536]
- 414.
- Luderer HF, Nazarian RM, Zhu ED, Demay MB. Ligand-dependent actions of the vitamin D receptor are required for activation of TGF-beta signaling during the inflammatory response to cutaneous injury. Endocrinology. 2013;154(1):16–24. [PMC free article: PMC3529380] [PubMed: 23132743]
- 415.
- Song L, Papaioannou G, Zhao H, Luderer HF, Miller C, Dall'Osso C, Nazarian RM, Wagers AJ, Demay MB. The Vitamin D Receptor Regulates Tissue Resident Macrophage Response to Injury. Endocrinology. 2016;157(10):4066–4075. [PMC free article: PMC5045513] [PubMed: 27526034]
- 416.
- Oda Y, Tu CL, Menendez A, Nguyen T, Bikle DD. Vitamin D and calcium regulation of epidermal wound healing. The Journal of steroid biochemistry and molecular biology. 2015 [PMC free article: PMC4753150] [PubMed: 26282157]
- 417.
- Chen S, Lewallen M, Xie T. Adhesion in the stem cell niche: biological roles and regulation. Development. 2013;140(2):255–265. [PMC free article: PMC3597204] [PubMed: 23250203]
- 418.
- Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437(7056):275–280. [PMC free article: PMC1399371] [PubMed: 16094321]
- 419.
- Li L, Hartley R, Reiss B, Sun Y, Pu J, Wu D, Lin F, Hoang T, Yamada S, Jiang J, Zhao M. E-cadherin plays an essential role in collective directional migration of large epithelial sheets. Cellular and molecular life sciences. CMLS. 2012;69(16):2779–2789. [PMC free article: PMC3459324] [PubMed: 22410739]
- 420.
- Bikle DD, Ng D, Tu CL, Oda Y, Xie Z. Calcium- and vitamin D-regulated keratinocyte differentiation. Mol Cell Endocrinol. 2001;177(1-2):161–171. [PubMed: 11377831]
- 421.
- van Etten E, Mathieu C. Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. The Journal of steroid biochemistry and molecular biology. 2005;97(1-2):93–101. [PubMed: 16046118]
- 422.
- Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. The Journal of pharmacology and experimental therapeutics. 2008;324(1):23–33. [PubMed: 17911375]
- 423.
- Gregori S, Casorati M, Amuchastegui S, Smiroldo S, Davalli AM, Adorini L. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. Journal of immunology. 2001;167(4):1945–1953. [PubMed: 11489974]
- 424.
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. [PubMed: 18510923]
- 425.
- Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC. DCs metabolize sunlight-induced vitamin D3 to 'program' T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8(3):285–293. [PubMed: 17259988]
- 426.
- Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. Journal of immunology. 2007;179(3):1634–1647. [PubMed: 17641030]
- 427.
- Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocrine reviews. 2008;29(6):726–776. [PMC free article: PMC2583388] [PubMed: 18694980]
- 428.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4(8):404–412. [PubMed: 18594491]
- 429.
- Adamopoulos IE, Bowman EP. Immune regulation of bone loss by Th17 cells. Arthritis Res Ther. 2008;10(5):225. [PMC free article: PMC2592787] [PubMed: 18983698]
- 430.
- Limketkai BN, Mullin GE, Limsui D, Parian AM. Role of Vitamin D in Inflammatory Bowel Disease. Nutr Clin Pract. 2017;32(3):337–345. [PubMed: 28537516]
- 431.
- Mathieu C, Van Etten E, Gysemans C, Decallonne B, Kato S, Laureys J, Depovere J, Valckx D, Verstuyf A, Bouillon R. In vitro and in vivo analysis of the immune system of vitamin D receptor knockout mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2001;16(11):2057-2065. [PubMed: 11697802]
- 432.
- O'Kelly J, Hisatake J, Hisatake Y, Bishop J, Norman A, Koeffler HP. Normal myelopoiesis but abnormal T lymphocyte responses in vitamin D receptor knockout mice. The Journal of clinical investigation. 2002;109(8):1091–1099. [PMC free article: PMC150940] [PubMed: 11956247]
- 433.
- Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, Steinman RM, Choi Y. TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. The Journal of experimental medicine. 2000;191(3):495–502. [PMC free article: PMC2195824] [PubMed: 10662795]
- 434.
- Baroni E, Biffi M, Benigni F, Monno A, Carlucci D, Carmeliet G, Bouillon R, D'Ambrosio D. VDR-dependent regulation of mast cell maturation mediated by 1,25-dihydroxyvitamin D3. J Leukoc Biol. 2007;81(1):250–262. [PubMed: 17035339]
- 435.
- Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Molecular endocrinology (Baltimore, Md). 2003;17(12):2386–2392. [PubMed: 14500760]
- 436.
- Gysemans C, van Etten E, Overbergh L, Giulietti A, Eelen G, Waer M, Verstuyf A, Bouillon R, Mathieu C. Unaltered diabetes presentation in NOD mice lacking the vitamin D receptor. Diabetes. 2008;57(1):269–275. [PubMed: 17959935]
- 437.
- Topilski I, Flaishon L, Naveh Y, Harmelin A, Levo Y, Shachar I. The anti-inflammatory effects of 1,25-dihydroxyvitamin D3 on Th2 cells in vivo are due in part to the control of integrin-mediated T lymphocyte homing. European journal of immunology. 2004;34(4):1068–1076. [PubMed: 15048717]
- 438.
- Wittke A, Weaver V, Mahon BD, August A, Cantorna MT. Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. Journal of immunology. 2004;173(5):3432–3436. [PubMed: 15322208]
- 439.
- Wittke A, Chang A, Froicu M, Harandi OF, Weaver V, August A, Paulson RF, Cantorna MT. Vitamin D receptor expression by the lung micro-environment is required for maximal induction of lung inflammation. Archives of biochemistry and biophysics. 2007;460(2):306–313. [PMC free article: PMC1933487] [PubMed: 17224129]
- 440.
- Adorini L. Intervention in autoimmunity: the potential of vitamin D receptor agonists. Cellular immunology. 2005;233(2):115–124. [PubMed: 15936743]
- 441.
- Ehrchen J, Helming L, Varga G, Pasche B, Loser K, Gunzer M, Sunderkotter C, Sorg C, Roth J, Lengeling A. Vitamin D receptor signaling contributes to susceptibility to infection with Leishmania major. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21(12):3208–3218. [PubMed: 17551101]
- 442.
- Rajapakse R, Mousli M, Pfaff AW, Uring-Lambert B, Marcellin L, Bronner C, Jeanblanc M, Villard O, Letscher-Bru V, Klein JP, Candolfi E. 1,25-Dihydroxyvitamin D3 induces splenocyte apoptosis and enhances BALB/c mice sensitivity to toxoplasmosis. The Journal of steroid biochemistry and molecular biology. 2005;96(2):179–185. [PubMed: 15939587]
- 443.
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt Rd R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–680. [PubMed: 12055625]
- 444.
- Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med. 2007;13(3):117–124. [PubMed: 17276732]
- 445.
- Schauber J, Gallo RL. The vitamin D pathway: a new target for control of the skin's immune response? Experimental dermatology. 2008;17(8):633–639. [PMC free article: PMC2729115] [PubMed: 18573153]
- 446.
- Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19(9):1067–1077. [PubMed: 15985530]
- 447.
- Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. Journal of immunology. 2004;173(5):2909–2912. [PubMed: 15322146]
- 448.
- Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, Helfrich YR, Kang S, Elalieh HZ, Steinmeyer A, Zugel U, Bikle DD, Modlin RL, Gallo RL. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. The Journal of clinical investigation. 2007;117(3):803–811. [PMC free article: PMC1784003] [PubMed: 17290304]
- 449.
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–1773. [PubMed: 16497887]
- 450.
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. The New England journal of medicine. 2002;347(15):1151–1160. [PubMed: 12374875]
- 451.
- Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24(3):341–348. [PubMed: 16546102]
- 452.
- Bilezikian JP, Bikle D, Hewison M, Lazaretti-Castro M, Formenti AM, Gupta A, Madhavan MV, Nair N, Babalyan V, Hutchings N, Napoli N, Accili D, Binkley N, Landry DW, Giustina A. MECHANISMS IN ENDOCRINOLOGY: Vitamin D and COVID-19. Eur J Endocrinol. 2020;183(5):R133–R147. [PMC free article: PMC9494342] [PubMed: 32755992]
- 453.
- Nogues X, Ovejero D, Pineda-Moncusi M, Bouillon R, Arenas D, Pascual J, Ribes A, Guerri-Fernandez R, Villar-Garcia J, Rial A, Gimenez-Argente C, Cos ML, Rodriguez-Morera J, Campodarve I, Quesada-Gomez JM, Garcia-Giralt N. Calcifediol Treatment and COVID-19-Related Outcomes. The Journal of clinical endocrinology and metabolism. 2021;106(10):e4017–e4027. [PMC free article: PMC8344647] [PubMed: 34097036]
- 454.
- Ustianowski A, Shaffer R, Collin S, Wilkinson RJ, Davidson RN. Prevalence and associations of vitamin D deficiency in foreign-born persons with tuberculosis in London. J Infect. 2005;50(5):432–437. [PubMed: 15907552]
- 455.
- Rook GA, Steele J, Fraher L, Barker S, Karmali R, O'Riordan J, Stanford J. Vitamin D3, gamma interferon, and control of proliferation of Mycobacterium tuberculosis by human monocytes. Immunology. 1986;57(1):159–163. [PMC free article: PMC1453883] [PubMed: 3002968]
- 456.
- Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. Journal of immunology. 2007;179(4):2060–2063. [PubMed: 17675463]
- 457.
- Sly LM, Lopez M, Nauseef WM, Reiner NE. 1alpha,25-Dihydroxyvitamin D3-induced monocyte antimycobacterial activity is regulated by phosphatidylinositol 3-kinase and mediated by the NADPH-dependent phagocyte oxidase. The Journal of biological chemistry. 2001;276(38):35482–35493. [PubMed: 11461902]
- 458.
- Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, Brennan PJ, Bloom BR, Godowski PJ, Modlin RL. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285(5428):732–736. [PubMed: 10426995]
- 459.
- Salahuddin N, Ali F, Hasan Z, Rao N, Aqeel M, Mahmood F. Vitamin D accelerates clinical recovery from tuberculosis: results of the SUCCINCT Study [Supplementary Cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis'. BMC Infect Dis. 2013;13:22. [PMC free article: PMC3556334] [PubMed: 23331510]
- 460.
- Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, Packe GE, Moore-Gillon JC, Darmalingam M, Davidson RN, Milburn HJ, Baker LV, Barker RD, Woodward NJ, Venton TR, Barnes KE, Mullett CJ, Coussens AK, Rutterford CM, Mein CA, Davies GR, Wilkinson RJ, Nikolayevskyy V, Drobniewski FA, Eldridge SM, Griffiths CJ. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet. 2011;377(9761):242–250. [PMC free article: PMC4176755] [PubMed: 21215445]
- 461.
- Schauber J, Oda Y, Buchau AS, Steinmeyer A, Zugel U, Bikle DD, Gallo RL. Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1,25-dihydroxyvitamin D3. The Journal of investigative dermatology. 2008;128(4):816–824. [PubMed: 17943182]
- 462.
- Norman AW, Roth J, Orci L. The vitamin D endocrine system: steroid metabolism, hormone receptors, and biological response (calcium binding proteins). Endocr Rev. 1982;3(4):331–366. [PubMed: 6295752]
- 463.
- Weishaar RE, Kim SN, Saunders DE, Simpson RU. Involvement of vitamin D3 with cardiovascular function. III. Effects on physical and morphological properties. Am J Physiol. 1990;258(1 Pt 1):E134–142. [PubMed: 2154115]
- 464.
- Walters MR, Ilenchuk TT, Claycomb WC. 1,25-Dihydroxyvitamin D3 stimulates 45Ca2+ uptake by cultured adult rat ventricular cardiac muscle cells. The Journal of biological chemistry. 1987;262(6):2536–2541. [PubMed: 3818607]
- 465.
- Selles J, Boland R. Rapid stimulation of calcium uptake and protein phosphorylation in isolated cardiac muscle by 1,25-dihydroxyvitamin D3. Mol Cell Endocrinol. 1991;77(1-3):67–73. [PubMed: 1816004]
- 466.
- Wu J, Garami M, Cao L, Li Q, Gardner DG. 1,25(OH)2D3 suppresses expression and secretion of atrial natriuretic peptide from cardiac myocytes. Am J Physiol. 1995;268(6 Pt 1):E1108–1113. [PubMed: 7611385]
- 467.
- Gardner DG, Chen S, Glenn DJ. Vitamin D and the heart. Am J Physiol Regul Integr Comp Physiol. 2013;305(9):R969–977. [PMC free article: PMC3840320] [PubMed: 24026071]
- 468.
- Boland R. Role of vitamin D in skeletal muscle function. Endocr Rev. 1986;7(4):434–448. [PubMed: 3536463]
- 469.
- Girgis CM, Clifton-Bligh RJ, Hamrick MW, Holick MF, Gunton JE. The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocrine reviews. 2013;34(1):33–83. [PubMed: 23169676]
- 470.
- Boland RL. VDR activation of intracellular signaling pathways in skeletal muscle. Mol Cell Endocrinol. 2011;347(1-2):11–16. [PubMed: 21664245]
- 471.
- Gelbard HA, Stern PH. U'Prichard DC. 1 alpha, 25-Dihydroxyvitamin D3 nuclear receptors in pituitary. Science. 1980;209(4462):1247–1249. [PubMed: 6250221]
- 472.
- Haug E, Gautvik KM. Demonstration and characterization of a 1 alpha,25-(OH)2D3 receptor- like macromolecule in cultured rat pituitary cells. J Steroid Biochem. 1985;23(5A):625–635. [PubMed: 3001410]
- 473.
- Tornquist K. Pretreatment with 1,25-dihydroxycholecalciferol enhances thyrotropin- releasing hormone- and inositol 1,4,5-trisphosphate-induced release of sequestered Ca2+ in permeabilized GH4C1 pituitary cells. Endocrinology. 1992;131(4):1677–1681. [PubMed: 1396313]
- 474.
- Tornquist K, Lamberg-Allardt C. The effect of 1,25-dihydroxy-cholecalciferol on the TRH induced TSH release in rats. Acta Endocrinol (Copenh). 1987;114(1):55–59. [PubMed: 3101341]
- 475.
- Narbaitz R, Sar M, Stumpf WE, Huang S, DeLuca HF. 1,25-Dihydroxyvitamin D3 target cells in rat mammary gland. Horm Res. 1981;15(4):263–269. [PubMed: 6100940]
- 476.
- Lopes N, Paredes J, Costa JL, Ylstra B, Schmitt F. Vitamin D and the mammary gland: a review on its role in normal development and breast cancer. Breast Cancer Res. 2012;14(3):211. [PMC free article: PMC3446331] [PubMed: 22676419]
- 477.
- Eisman JA, Suva LJ, Martin TJ. Significance of 1,25-dihydroxyvitamin D3 receptor in primary breast cancers. Cancer Res. 1986;46(10):5406–5408. [PubMed: 3019526]
- 478.
- Eisman JA, Sutherland RL, McMenemy ML, Fragonas JC, Musgrove EA, Pang GY. Effects of 1,25-dihydroxyvitamin D3 on cell-cycle kinetics of T 47D human breast cancer cells. J Cell Physiol. 1989;138(3):611–616. [PubMed: 2925799]
- 479.
- Colston KW, Berger U, Coombes RC. Possible role for vitamin D in controlling breast cancer cell proliferation. Lancet. 1989;1(8631):188–191. [PubMed: 2563099]
- 480.
- Duncan WE, Whitehead D, Wray HL. A. 1,25-dihydroxyvitamin D3 receptor-like protein in mammalian and avian liver nuclei. Endocrinology. 1988;122(6):2584–2589. [PubMed: 2836167]
- 481.
- Cao Y, Shu XB, Yao Z, Ji G, Zhang L. Is vitamin D receptor a druggable target for non-alcoholic steatohepatitis? World J Gastroenterol. 2020;26(38):5812–5821. [PMC free article: PMC7579753] [PubMed: 33132636]
- 482.
- Rixon RH, MacManus JP, Whitfield JF. The control of liver regeneration by calcitonin, parathyroid hormone and 1 alpha,25-dihydroxycholecalciferol. Mol Cell Endocrinol. 1979;15(2):79–89. [PubMed: 499650]
- 483.
- Nguyen TM, Guillozo H, Marin L, Dufour ME, Tordet C, Pike JW, Garabedian M. 1,25-dihydroxyvitamin D3 receptors in rat lung during the perinatal period: regulation and immunohistochemical localization. Endocrinology. 1990;127(4):1755–1762. [PubMed: 2169401]
- 484.
- Gaultier C, Harf A, Balmain N, Cuisinier-Gleizes P, Mathieu H. Lung mechanics in rachitic rats. Am Rev Respir Dis. 1984;130(6):1108–1110. [PubMed: 6508008]
- 485.
- Herscovitch K, Dauletbaev N, Lands LC. Vitamin D as an anti-microbial and anti-inflammatory therapy for Cystic Fibrosis. Paediatr Respir Rev. 2014;15(2):154–162. [PubMed: 24332502]
- Review Disorders in the Action of Vitamin D.[Endotext. 2000]Review Disorders in the Action of Vitamin D.Liberman U, Bikle DD. Endotext. 2000
- Review Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System.[Nutrients. 2022]Review Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System.Latic N, Erben RG. Nutrients. 2022 Dec 6; 14(23). Epub 2022 Dec 6.
- Vitamin D Receptor Mediates a Myriad of Biological Actions Dependent on Its 1,25-Dihydroxyvitamin D Ligand: Distinct Regulatory Themes Revealed by Induction of Klotho and Fibroblast Growth Factor-23.[JBMR Plus. 2021]Vitamin D Receptor Mediates a Myriad of Biological Actions Dependent on Its 1,25-Dihydroxyvitamin D Ligand: Distinct Regulatory Themes Revealed by Induction of Klotho and Fibroblast Growth Factor-23.Haussler MR, Livingston S, Sabir ZL, Haussler CA, Jurutka PW. JBMR Plus. 2021 Jan; 5(1):e10432. Epub 2020 Dec 3.
- Review Role of vitamin D, its metabolites, and analogs in the management of osteoporosis.[Rheum Dis Clin North Am. 1994]Review Role of vitamin D, its metabolites, and analogs in the management of osteoporosis.Bikle DD. Rheum Dis Clin North Am. 1994 Aug; 20(3):759-75.
- Review Vitamin D physiology.[Prog Biophys Mol Biol. 2006]Review Vitamin D physiology.Lips P. Prog Biophys Mol Biol. 2006 Sep; 92(1):4-8. Epub 2006 Feb 28.
- Vitamin D: Production, Metabolism and Mechanisms of Action - EndotextVitamin D: Production, Metabolism and Mechanisms of Action - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...