INTRODUCTION
Asbestos is a term applied to several mineral species when they occur in a fibrous form (asbestiform). When the mineral species are asbestiform, they have the physical characteristics associated with asbestos, such as large aspect ratio of fibers, flexibility, separability and weavability of fibers, and chemical and physical durability. However, in addition to those common properties, each asbestos mineral species has unique chemical and physical properties that make it distinct from the others. Details about the nature and limitations of techniques used to identify and characterize asbestos fibers will not be discussed here, but can be found in reference sources such as Spurny (1994) and Roggli et al. (1992).
This chapter provides an overview of asbestos mineralogy, focusing on characteristics of asbestos fibers that are potentially relevant to carcinogenicity. In particular, the various asbestos mineral species are described, with an emphasis on the characteristics and properties related to their unique biologic properties. Minerals are known to interact dynamically with their environment particularly when they are in contact with a fluid. Such interactions often occur at the interface between the mineral and its environment, in other words, at the mineral’s surface. These interactions are critically important in many natural environments and include such phenomena as dissolution and precipitation (which alter the fluid’s composition), oxidation and reduction of species in the fluid, sorption, and ion exchange. Each of those phenomena has a potential role in mineral-induced pathogenesis, including carcinogenesis and fibrosis, although understanding of the relationship between mineralogic properties and pathogenesis remains incomplete.
The concept of mineral species is fundamental to mineralogy. A mineral species is a crystalline solid with a specific atomic structure and a specific chemical composition (or compositional range). The specific crystal structure and chemical composition of each mineral species imparts a unique set of properties, including how the species interacts physically and chemically with its environment. In a system paralleling that for the plant and animal kingdoms, mineral species are classified hierarchically. A mineral group is roughly equivalent to the family classification and consists of minerals with similar compositions or structures. Minerals may also exhibit variability within a species with respect to a particular property. For example, some mineral species may occur with an asbestiform habit (physical form) or a non-asbestiform habit. Those are typically not given distinct mineral-species names but instead are referred to as varieties of the same species; sometimes, they are given varietal names, as in the case of crocidolite, which is the asbestiform variety of the mineral species riebeckite.
Other mineral groups may have species with occasional asbestiform varieties, but the primary mineral groups for asbestos are amphibole and serpentine. Each species of these groups has a distinct crystal structure, but chemical compositions vary between species within the group. The principal mineral species constituting asbestos are detailed below; they include asbestiform serpentine (chrysotile) and asbestiform varieties of amphibole, such as tremolite, actinolite, anthophyllite, grunerite, riebeckite (also known as crocidolite), winchite, and richterite. Table 3.1 lists the mineral species, varietal names, and mineral groups associated with the common asbestos minerals. Although the three chrysotile mineral species all have the same ideal chemical formula, these polymorphs (or polytypes) differ in the nature of the stacking relationship between successive layers, with clinochrysotile being the most abundant type (Gaines et al. 1997).
TABLE 3.1
Asbestos Minerals.
“FIBROUS” AND “ASBESTIFORM”
Many minerals may occur as small particles, including particles in the respirable size range, which is less than about 10 μm in aerodynamic diameter. Of these, some may include particles with aspect ratios (length: diameter) of 5:1 or more, usually reflecting a characteristic of the underlying crystal structure. For example, asbestiform amphiboles have fibers that are elongate parallel to the underlying silicate chains in the structure.
Fibrous is a term applied to minerals that consist of fibers, that is, exhibit a large aspect ratio. Although the minimal aspect ratio of a mineral fiber may be debated, for the purpose of definition observed aspect ratios in general are very large (for example, over 5:1 and sometimes over 100:1).
Asbestiform refers to a subset of fibrous minerals. Among fibrous minerals, some exhibit the additional qualities of flexibility and separability (which contribute to weavability). Such minerals are referred to as asbestiform. Typically, asbestiform minerals also have relatively small fiber diameters (usually under 1 μm) and large fiber lengths (such as 5-10 μm). The asbestiform characteristics are related to properties of the underlying crystal structures, with the specific relationship according to the mineral group. For example, it has been suggested that flexibility is related to defects in the crystal structure of the asbestiform varieties of amphibole (Veblen and Wylie 1993), whereas flexibility in asbestiform serpentine (the various forms of chrysotile) may be related to the hydrogen bonding between concentric sheets of 1:1 layers, as described below.
Some mineral species have both asbestiform and non-asbestiform varieties, and these varieties may have properties beyond just their flexibility that differ. For example, consider the grain boundaries in asbestiform amphibole. Asbestos fibers typically occur as parallel bundles of fibrils (filaments consisting of individual crystals) that are bound together along grain boundaries. The material along the grain boundaries typically is not am phibole but rather a layer silicate, such as talc or mica. When the material is processed, fibers are produced by the breaking apart of packets of fibrils by separation along the structurally weaker grain boundaries, which allows the layer-silicate material to become the surface of the fiber. It is this crystalline material that interacts with the biologic system after inhalation or ingestion. In contrast, the surface of a non-asbestiform variety of amphibole (either an acicular crystal or a cleavage fragment) is often amphibole (and not layer silicate) because the particles are formed either by growth of the original amphibole crystal in the case of acicular fibers or by fracture along weaker atomic planes in the amphibole structure. Hence, asbestiform amphibole is likely to have a different surface structure and composition from non-asbestiform amphibole. Those differences in surface material result in different surface properties between asbestiform and non-asbestiform minerals of the same species, which may in turn result in different biologic responses.
Some fibrous but non-asbestiform minerals also pose potential concern with respect to human exposure. For example, the fibrous zeolite erionite has been associated with human cases of mesothelioma after environmental exposure (Baris et al. 1987).
SERPENTINE ASBESTOS (CHRYSOTILE) MINERALOGY
Chrysotile—sometimes called white asbestos—is the most common type of asbestos to be used commercially, accounting for about 85% of world asbestos production in 1977 (Liddell 1997, Schreier 1989). At present, chrysotile is the only type of asbestos used in manufacturing in the United States (ATSDR 2001). In addition, chrysotile and other serpentine minerals are common naturally, particularly in hydrothermally altered, magnesium-rich rocks, such as altered basalt, peridotite, and dunite. Many such rocks have been almost completely altered to serpentine and are referred to as serpentinites. Although lizardite is the most common form of serpentine in these rocks, chrysotile can also be present, typically having formed as a late-stage mineral filling veins and sometimes replacing the bulk rock. Chrysotile has been commercially exploited in Canada (Quebec and Ontario), the United States (Vermont and California), Zimbabwe, Russia, South Africa, Australia, and elsewhere (Ross 1981), and it has been used in various products, including insulation, friction materials (such as brake pads), and fiber-reinforced composites (such as concrete) (Harrison et al. 1999, Ross and Virta 2001). In addition to synthetic chrysotile-bearing materials, natural deposits are possible sources of exposure to chrysotile, either by direct exposure to chrysotile-bearing rocks and soils or by redistribution of chrysotile fibers from large natural deposits, such as occurs at Coalinga, California (Klein 1993). It has been argued that atmo spheric processes have redistributed Coalinga chrysotile over the entire Northern Hemisphere from its occurrence in soils in a 50-mi2 area (Klein 1993).
Serpentine minerals belong to a family of 1:1 layer silicates, which are composed of a sheet of polymerized SiO44− tetrahedra (with silicon at the center of each tetrahedron and oxygen at each apex) that is bonded to a sheet of polymerized Mg(OH)64− octahedra (with magnesium at the center of each octahedron and oxygen at each apex) (Figure 3.1). This ratio of tetrahedral to octahedral sheets gives the 1:1 layer silicates their name. The tetrahedral:octahedral (1:1) polymerized layers are stacked one atop another to form the chrysotile structure.
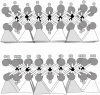
FIGURE 3.1
Lizardite structure viewed down the a-axis. Polymerized silica tetrahedra form a sheet at the bottom of each 1:1 unit (two units are shown stacked vertically), and magnesium hydroxide octahedra form a sheet drawn as ball-and-stick. In chrysotile, the (more...)
The serpentine group is based on a metal hydroxide sheet containing Mg2+ cations, giving rise to a composition of Mg3Si2O5(OH)4.
Chrysotile exhibits a smaller variation in chemical composition than other (non-asbestiform) serpentine minerals, but substitutions do occur. The most common substitutions are Si4+→Al3+, Mg2+→Fe2+, and Mg2+→Al3+; however, these substitutions typically represent much less than 10% of the atomic sites (Veblen and Wylie 1993). Other metal substitutions (such as Ni, Co, Mn, Cr, and Zn) may occur in trace amounts (Ross 1981).
Dimensionally, the octahedral (Mg) sheet is slightly larger than the tetrahedral (Si) sheet. The two sheets are bonded to one another by the sharing of some of their oxygen atoms; the natural spacing of the atoms in the octahedral sheet is about 3.6% larger than the natural spacing in the tetrahedral sheet (Veblen and Wylie 1993). This structural mismatch can be accommodated either by a curving of the layers (as first proposed by Linus Pauling in 1930 on theoretical grounds) by cation substitution. In chrysotile, layer curvature exposes the magnesium octahedral sheet at the fiber surface, thereby reducing strain from the dimensional mismatch. Whittaker (1957) calculated the strain-free diameter for a single chrysotile fiber on the basis of a pure Mg octahedral sheet; his value of 0.02 μm compares favorably with particle diameters measured from real samples (0.03-0.17 μm), as reported by Veblen and Wylie (1993). The particles measured in the studies reported by Veblen and Wylie may consist of multiple fibers. In natural samples of chrysotile, some of the strain may also be relieved by cation substitution, which allows the particles to achieve slightly larger diameters (Gaines et al. 1997). Cation substitution in chrysotile is typically more limited than in the other magnesium-serpentine minerals (lizardite and antigorite; chrysotile’s composition is closer to the ideal Mg3Si2O5(OH)4.
Dissolution of chrysotile is likely to occur after contact with physiologic fluids. The kinetics of chrysotile dissolution have been studied extensively in experimental systems. Dissolution in the mid pH range (4-7) appears to be independent of pH (Hume and Rimstidt 1992), with Mg2+ release occurring more rapidly initially than silica release but leveling off after at most a few atomic layers of material have been removed, as consistent with the data presented in Hume (1991). At 37°C and under ionic strengths similar to those in lung fluids, Hume and Rimstidt (1992) measured a dissolution rate (k) of 5.9×10−10 mol m−2 sec−1. At lower pH, the rate would be expected to increase substantially, but no comprehensive quantitative study has been done on chrysotile dissolution rate as a function of pH in acidic environments. At the stated rate, a chrysotile particle, even as thick as 1 μm, would be predicted to be removed from the lung by dissolution in less than a year. The process would remove the pathogenic par ticle, but it would also release into the surrounding environment any trace metals from the particle, which could be toxic in their own right, although probably in a transient fashion.
AMPHIBOLE ASBESTOS MINERALOGY
Amphiboles are common silicate minerals found in many types of rocks. Although most occurrences of amphibole are non-asbestiform, large deposits of some asbestiform amphiboles have been exploited commercially, particularly from deposits in South Africa, Australia, and Finland. Those that have been exploited commercially typically belong to a small subset of amphibole mineral species (riebeckite or crocidolite, grunerite, anthophyllite, actinolite, and tremolite). As discussed below, other amphiboles may also occur with asbestiform habits, including winchite and richterite, which are associated with human exposures in Libby, Montana. In addition, some amphiboles occur with fibrous but non-asbestiform habits, such as byssolite (the stiff-fibered form of actinolite).
Amphibole minerals form a family of double-chain silicates, which are composed of I-beams, as shown in Figure 3.2. Chains of polymerized silica tetrahedra are on both the top and bottom of the I-beam. Between the silica chains lies a sheet of octahedrally coordinated metal ions (specifically, the atomic B sites). At the midpoints along the edges of the I-beam are the eight slightly larger and coordinated C sites. The I-beams—which run the length of the fibers in asbestiform amphibole—are stacked as shown in Figure 3.2c, creating an additional atomic site, the A site.

FIGURE 3.2
Amphibole structure. a) Individual I-beam down a-axis, showing two chains of polymerized silica tetrahedra (one darker) overlying strip of metal octahedra (shown as ball-and-stick). A sites are in channels formed by stacked I-beams; B sites appear as (more...)
The complexity of atomic sites in amphiboles is reflected in their chemi cal complexity. The general formula for amphiboles can be described as A0−1B2C5[T8O22](OH)2, where A sites are coordinated by 12 oxygens and can accommodate large monovalent cations; B sites are coordinated by eight oxygens and can accommodate large monovalent and divalent cations; C sites are coordinated by six oxygens and can accommodate only smaller cations; and T sites are four coordinated tetrahedral sites that can accommodate only very small cations, such as Si4+ and Al3+. Table 3.2 lists compositions of amphiboles that have been recognized in association with disease in humans. The compositions shown are idealized chemical formulas, but natural samples often have compositions that differ slightly with respect to both major and trace elements. For example, analysis of the amphibole from Libby, Montana, that has been associated with respiratory disease (Gunter et al. 2003) shows substantial deviation from the ideal chemical formulae for either winchite or richterite because of substitutions of isovalent cations or anions or because of a portion of atomic sites being empty:
TABLE 3.2
Mineral Names, Varietal Names, and Atomic Site Compositions for Amphiboles That Have Been Commonly Encountered in an Asbestiform Habit.
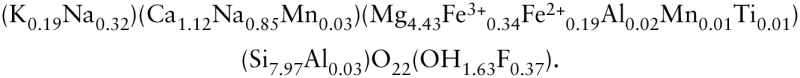
Dimensionally, amphibole asbestos fibers exhibit a range of diameters. As reported in Veblen and Wylie (1993), particle diameters as measured on both bulk and airborne samples fall in the range 0.06-0.70 μm, with riebeckite (crocidolite) particles generally thinner and anthophyllite asbestos particles thicker.
Amphibole dissolution is considerably slower than chrysotile dissolution, as measured experimentally. Although no rates have been reported specifically for dissolution of asbestiform amphibole, experimentally determined rates for amphiboles in general typically fall in the range of 10−12-10−10 mol m−2 sec−1, that is, orders of magnitude smaller than those for chrysotile. Such dissolution rates imply that a typical amphibole fiber will not dissolve in the lung over the course of a human lifetime. In fact, amphibole fibers often serve as sites of precipitation in the lung, becoming coated with iron-rich material to form an asbestos body. Whether amphiboles would dissolve substantially in lower-pH physiologic fluids, as would be found in the stomach, is not known.
PROPERTIES OF POTENTIALLY HAZARDOUS FIBROUS MINERALS
Several physical and chemical factors may contribute to a mineral particle’s pathogenic potential (Table 3.3). Many properties are related to how a mineral interacts with a fluid under various conditions. The impor tance of those properties in natural environments is well recognized, but they have not been studied in the context of the pathogenesis of cancer or other diseases by minerals. They are discussed here to provide a context for the potential roles of minerals in biologic processes that lead to disease. Their potential role in pathogenesis will be discussed below. Crystal structure and composition determine a mineral’s properties, so each of the different mineral species discussed above will behave somewhat differently as it interacts with body fluids.
TABLE 3.3
Mineralogic Properties in Relation to Pathogenesis.
Particle size and shape are widely recognized to be important in determining the deposition and translocation of a particle, particularly in the context of respirable particles (e.g., Lehnert 1993). In general, the smaller the particle, the further it can be transported before settling because of gravitational forces. The net effect is that particles with aerodynamic diameters less than about 5 μm are more likely to reach the lower airways, whereas larger particles are usually deposited higher in the respiratory tract and perhaps cleared via the mucociliary escalator. Consequently, the distribution of particle sizes and shapes to which a particular potential target organ or site is exposed may differ from the distribution of the dose to which another site is exposed (Quinn et al. 1997). Different particle size-shape populations may, in turn, have of different physical or chemical characteristics. Two important factors are directly associated with particle size: surface area per mass and size relative to cells. The first factor affects surface-controlled reactions, including many of those discussed below; surface-controlled reactions are those that occur between a mineral surface and its environment (for example, a physiologic fluid), such as dissolution, sorption, and oxidation-reduction. The second factor influences how a cell interacts with a particle, which is most important when a particle is roughly cell-sized or smaller. For example, macrophages have been observed to attempt phagocytosis of particles (fibers in particular) that are around 5-10 μm in length or greater; this process can result in the release of inflammatory agents to the local environment. When particles are much smaller than a macrophage (about 1-2 μm), they are readily cleared via phagocytosis.
Mineral dissolution has several potential roles in the biologic response to exposure to asbestos. One role is to remove a particle. Of the minerals discussed here, only chrysotile is expected to dissolve substantially under most physiologic conditions, and the potential for chrysotile fibers to dissolve while amphibole fibers are far more persistent may be relevant to the relative carcinogenicity of the two main fiber types. Hume and Rimstidt (1992) estimated lifetimes of about 9 months for a 1-μm-diameter fiber, and their model predicts that fibers with diameters of 0.1 μm would require only weeks to dissolve completely. As a mineral dissolves, material is removed from its surface, and this can affect structure and composition and therefore surface properties. Hence, mineral dissolution may affect any pathogenic process that is related to surface interactions (such as those discussed below). Another potential role for dissolution in pathogenesis is the release of trace elements from the crystal structure. In particular, some of the minerals discussed contain trace amounts of polyvalent cations that could have a role in mineral-induced pathogenesis. Such a process has been related to the observed high potency of iron-bearing asbestos minerals in some experimental systems. For example, it has been postulated that iron released from some types of asbestos by dissolution serves as an oxidation-reduction catalyst in a Fenton-type reaction to produce free radicals (Aust and Lund 1990).
Mineral precipitation can occur when the concentration of dissolved aqueous species reaches a critical value that depends on the mineral species. Mineral precipitation has been observed to occur on the surface of some asbestos fibers after a period of time, forming particles known as asbestos bodies or ferruginous bodies. Although the details of formation are not fully known, an asbestos body reflects the precipitation of ferric iron hydroxides on a particle’s surface, consequently radically changing the surface properties of the particle. The native asbestos surface is no longer exposed, but instead the surface may consist of a quasi-crystalline material with a high sorption capacity and with relatively low solubility (the solubility of ferric iron, Fe3+, is much lower than the solubility of ferrous iron, Fe2+). Ferruginous coatings have also been observed to form on minerals other than asbestos (Roggli et al. 1992).
Sorption is the process by which atoms or molecules in a fluid bind to the surface of a mineral. Sorption has numerous roles in mineral-fluid interactions, including being a component of many of the processes described here (dissolution, precipitation, oxidation-reduction, and acid-base catalysis). It can also affect reactions in a fluid by allowing atoms and molecules to be concentrated at the mineral surface, thereby effectively raising their activities. Furthermore, sorption processes often orient molecules on the basis of the stereochemistry of the surface and the molecules. Those two aspects of sorption can initiate reactions among fluid species that might otherwise proceed slowly or not at all; for example, Ferris and Ertem (1992) found that the clay mineral montmorillonite catalyzes the oligomerization of ribonuleotides from aqueous solution. Finally, sorption of hazardous molecules (perhaps before introduction of a particle to the physiological environment) can allow the particle to function as an effective delivery agent; this has been suggested to be the case for mineral sorption of constituents from cigarette smoke or diesel exhaust.
Ion exchange is the process by which cations (typically monovalent or divalent cations, such as Na+, K+, and Ca2+) that are loosely bound to a mineral are able to exchange with a monovalent or divalent cation in solution. The exchange capacity of the mineral is related to the proportion of exchangeable cation sites that are directly accessible by the fluid (the surface sites) or by cations along rapid-diffusion pathways (such as channels greater than about 0.3 nm); hence, a particle has a finite potential for ion exchange. Nevertheless, ion exchange can effectively buffer the activity of a cation at the surface of a particle for some time. Because of their high surface-area-to-mass ratio, small asbestos particles could have a relatively large ion exchange capacity. Particularly for amphibole fibers with ions in the A sites (such as crocidolite), ion activity is important in many cellular signaling pathways, but whether mineral-induced ion exchange can affect these pathways remains to be investigated.
Mineral surfaces may be the site of catalytic functionalities, such as proton or electron donors and acceptors. Proton donor or acceptor sites can function as acid-base catalysts in many reactions. Numerous types of such sites are present on mineral surfaces (Hochella 1993). In freshly fractured silicate minerals (including asbestos), broken bonds result in surface silica sites that are unde saturated with respect to ionic charge. As those sites interact with water and dissipate the surface charge, they generate free radicals in the fluid. Fubini et al. (1990) have shown that the process can be particularly important in silica-induced pathogenesis. Ultimately, the sites become silanol groups (Si-OH) that will protonate or deprotonate in response to pH. Indeed, many surface oxygens will function similarly, and the acidic strength of protons on these sites depends on the local charge distribution in the underlying mineral.
Surface-induced oxidation-reduction is another catalytic pathway for mineral surfaces. Oxidation-reduction involves the exchange of electrons between the mineral surface and a fluid species; it results in the oxidation of the mineral site and reduction of the fluid species, or vice versa. Such processes are observed in natural environments; the mineral surface donates electrons and thereby reduces species in the fluid and commonly forms metal precipitation at structurally determined sites (Ilton et al. 1992). That process can occur in minerals that contain polyvalent cations (such as Fe2+ and Fe3+) in sites that are sufficiently close to allow charge transfer (as in the case of the octahedral strips in amphiboles). In fact, under some pH regimes, mineral surfaces are stronger redox catalysts than iron in solution (Hochella 1993); this suggests that the Fenton reaction proposed for free-radical formation by some asbestos may be as likely with iron on the mineral surface as with dissolved iron in the fluid. Although that process has not been investigated directly in relation to mineral-induced pathogenesis, indirect observations support the idea that it is important in physiologic fluids. For example, observations of mineral particles recovered from the lung show micas with precipitation on the edges consistent with a structurally controlled reduction-precipitation process (Roggli et al. 1992).
REFERENCES
- ATSDR (Agency for Toxic Substances and Disease Registry). Toxicological Profile for Asbestos. Atlanta, GA: US Department of Health and Human Services; 2001.
- Aust AE, Lund LG. The role of iron on asbestos-catalyzed damage to lipids and DNA. In: Reddy CC, Hamilton GA, Madyastha KM, editors. Biological Oxidation Systems. 1-2. San Diego, CA: Academic Press; 1990. pp. 597–606.
- Baris I, Simonato L, Artvinli M, Pooley F, Saracci R, Skidmore J, Wagner C. Epidemiological and environmental evidence of the health effects of exposure to erionite fibres: A four-year study in the Cappadocian region of Turkey. International Journal of Cancer. 1987;39(1):10–17. [PubMed: 3025107]
- Ferris JP, Ertem G. Oligomerization of ribonucleotides on montmorillonite: Reaction of the 5'-phophorimidazolide of adenosine. Science. 1992;257:1387–1389. [PubMed: 1529338]
- Fubini B, Giamello E, Volante M, Bolis V. Chemical functionalitites at the silica surface determine its reactivity when inhaled formation and reactivity of surface radicals. Toxicology and Industrial Health. 1990;6(6):571–598. [PubMed: 1965871]
- Gaines RV, Skinner HCW, Ford EE, Mason B, Rosenzweig A. Dana’s New Mineralogy. New York: John Wiley and Sons; 1997. p. 1810.
- Gunter ME, Dyar MD, Twamley B, Foit F, Cornelius S. Composition, Fe3+/SFe, and crystal structure of non-asbestiform and asbestiform amphiboles from Libby, Montana, USA. American Mineralogist. 2003;88:1970–1978.
- Harrison PTC, Levy LS, Patrick G, Pigott GH, Smitt LL. Comparative hazards of chrysotile asbestos and its substitutes: A European perspective. Environmental Health Perspectives. 1999;107:607–612. [PMC free article: PMC1566482] [PubMed: 10417355]
- Hochella MF. Surface chemistry, structure, and reactivity of hazardous mineral dust. In: Guthrie GD, Mossman BT, editors. Health Effects of Mineral Dusts. Vol. 28. Mineralogical Society of America; 1993. pp. 275–308. Reviews in Mineralogy.
- Hume LA. The Dissolution of Chrysotile. Blacksburg, VA: Virginia Polytechnic Institute and State University; 1991. p. 46. M.S. thesis.
- Hume LA, Rimstidt JD. The biodurablity of chrysotile asbestos. American Mineralogist. 1992;77:1125–1128.
- Ilton ES, Earley D, Marozas D, Veblen DR. Reaction of some trioctahedral micas with copper-sulfate solutions at 25°C and 1 atomsphere: An electron microphobe and transmission electron microscopy investigation. Economic Geology. 1992;87:1813–1829.
- Klein C. Rocks, minerals, and a dusty world. Reviews in Mineralogy. 1993;28:6–138.
- Lehnert BE. Defense mechanisms against inhaled particles and associated particle-cell interactions. Reviews in Mineralogy and Geochemistry. 1993;28:427–469.
- Liddell F. Magic, menace, myth and malice. Annals of Occupational Hygiene. 1997;41(1):3–12. [PubMed: 9072948]
- Mellini M. The crystal structure of lizardite 1T: Hydrogen bonds and polytypism. American Mineralogist. 1982;67:587–598.
- Papike JJ, Ross M, Clark JR. Crystal chemical characterization of clinoamphiboles based on five new structure refinements. Mineralogical Society of America Special Paper. 1969;2:117–137.
- Pauling L. The structure of the chlorites. Proceedings of the National Academy of Sciences of the United States of America. 1930;16:578–582. [PMC free article: PMC526695] [PubMed: 16587609]
- Quinn MM, Ellenbecker MJ, Smith TJ, Wegman DH, Eisen EA. A model to predict deposition of man-made vitreous fibres in the human tracheobronchial region. Annals of Occupational Hygiene. 1997;41 (Supplement 1):197–202.
- Roggli VL, Pratt PC, Brody AR. Analysis of tissue mineral fiber content. In: Roggli V, Greenburg S, Pratt P, editors. Pathology of Asbestos-Associated Diseases. 1st edition. New York: Lippincott Williams and Wilkins; 1992. pp. 305–354.
- Ross M. The geologic occurrences and health hazards of amphibole and serpentine asbestos. Reviews in Mineralogy and Geochemistry. 1981;9A:279–323.
- Ross M, Virta R. Occurrence, production and uses of asbestos. Canadian Mineralogist. 2001;5:79–88.
- Schreier H. Asbestos in the Natural Environment. New York: Elsevier Science Publication; 1989.
- Spurny KR. Sampling, analysis, identification, and monitoring of fibrous dusts and aerosols. The Analyst. 1994;119:41–51.
- Veblen DR, Wylie AG. Mineralogy of amphiboles and 1:1 layer silicates. Reviews in Mineralogy and Geochemistry. 1993;28:61–138.
- Whittaker EJW. The structure of chrysotile. V. Diffuse reflexions and fibre texture. ACTA Crystallographica. 1957;10 (Part 3):149–156.
Publication Details
Copyright
Publisher
National Academies Press (US), Washington (DC)
NLM Citation
Institute of Medicine (US) Committee on Asbestos: Selected Health Effects. Asbestos: Selected Cancers. Washington (DC): National Academies Press (US); 2006. 3, Background Information on Asbestos.

