

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Selective inhibitors of FAS-TE
Robert Ardecky, Michael P. Hedrick, Palak Gosalia, Fusayo Yamamoto, Monika Milewski, Nikki Barron, Qing Sun, Jiwen Zhou, Santhi Ganji, Alka Mehta, Elliot Sugarman, Kevin Nguyen, Stefan Vasile, Eigo Suyama, Peter Teriete, Douglas Sheffler, Pooi San Lee, Margrith Mattmann, Jessica De Ingeniis, Derek Stonich, Arianna Mangravita-Novo, Sumeet Salaniwal, Paul Kung, Jena Diwan, Layton H. Smith, Eduard Sergienko, Thomas D.Y. Chung, Anthony B. Pinkerton, Nicholas D. P. Cosford, and Jeffrey W. Smith.
Author Information and Affiliations
Received: April 12, 2013; Last Update: January 13, 2014.
Fatty acid synthase (FAS) is the single human enzyme that can convert dietary carbohydrate to fat. The FAS protein contains six enzymatic domains and an acyl-carrier protein (ACP). The final enzymatic pocket is a thioesterase (TE), which liberates the final product (palmitate) from its link to the ACP. Notably, FAS has only marginal importance in adults because dietary fat provides for normal physiology. However, a link between FAS and cancer was discovered in 1994 when Frank Kuhajda found that the OA-519 antigen, a marker for poor prognosis in breast and prostate cancer, is actually fatty acid synthase. A number of subsequent immunohistochemical analyses showed that increased expression of FAS is a hallmark of all major cancers including those of the prostate, the breast, the colon, and the ovaries. Therefore, FAS represents a compelling target for anticancer drug discovery. To our knowledge, no thioesterase (TE) has ever been targeted for drug development, including the TE domain of FAS.
This report describes the discovery of a series of aminothiazoles as selective FAS-TE inhibitors. ML356, the lead compound from this series, is potent (IC50 = 0.334 μM) and selective inhibitor of FAS-TE. The compounds are selective for FAS over a number of other human thioesterases expressed in tumor cells, and selective for the thioesterase domain of FAS over its most closely related human homolog, ACOT4. ML356 also blocks the biosynthesis of palmitate (the end product of FAS) in cells.
Assigned Assay Grant #: DA032474-01 (derived from 1 R01 CA140427-01A1 Cycle 18 - Fast Track)
Screening Center Name & PI: Sanford-Burnham Medical Research Institute & John C. Reed, M.D., Ph.D.
Chemistry Center Name & PI: Sanford-Burnham Medical Research Institute & John C. Reed, M.D., Ph.D.
Assay Submitter & Institution: Jeffrey Smith, Ph.D., Sanford-Burnham Medical Research Institute
PubChem Summary Bioassay Identifier (AID): 602265
Probe Structure & Characteristics
This Center Probe Report describes ML356, a selective inhibitor of FAS-TE. ML356 has a central aminothiazole core. The chemical structure and data summary are shown in Table 1.
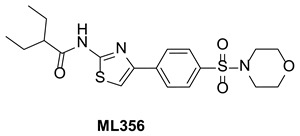
Table 1Summary of Probe ML356 Characteristics
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID] |
|---|---|---|
| CID 70680643 ML356 | FAS-TE Inhibitor Fatty Acid Synthase (Thioester domain) | 334 ± 0.03 (n=4) SID 160658425, AID 652164 |
1. Recommendations for Scientific Use of the Probe
The FAS protein contains six enzymatic domains and an acyl-carrier protein (ACP). The final enzymatic pocket is a thioesterase, which liberates the final product (palmitate) from its link to the ACP. It is the thioesterase domain of FAS, which we plan to target here. To our knowledge, no thioesterase (TE) has ever been targeted for drug development. Consequently, the unique enzymatic target and the new chemical entities that will be developed are one novel aspect of the study. Another novel aspect of the study is our intent to identify selective FAS inhibitors, by counter screening against other thioesterases.
This study will focus on developing drug-like inhibitors/probes against FAS, an enzyme that is essential for growth of solid tumors. Notably, FAS has only marginal importance in adults because dietary fat provides for normal physiology. The link between FAS and cancer was discovered in 1994 when Frank Kuhajda found that the OA-519 antigen, a marker for poor prognosis in breast and prostate cancer [1], is actually fatty acid synthase [2]. A number of subsequent immunohistochemical analyses showed that increased expression of FAS is a hallmark of all major cancers (for review see [3]), including those of the prostate [4-6], the breast [7,8], the colon [9,10], and the ovaries [11,12].
Perhaps more significantly, increased expression of FAS in tumors is usually linked with poor prognosis. FAS is an indicator of poor prognosis in cancers of the prostate [13], ovaries [11], and colon [14]. In breast cancer, FAS is a predictor of disease recurrence [8], whereas the lack of FAS predicts freedom from disease and overall survival [8]. FAS is also linked to poor prognosis in soft tissue sarcomas [6], Wilms tumor [15], renal cell carcinoma [16], and squamous cell carcinoma of the tongue [17]. The correlation between expression of FAS and poor prognosis strongly suggests that this enzyme is mechanistically linked to disease progression, providing a strong rationale for pursuing the development of FAS inhibitors.
Indeed, many other studies show that the requirement for FAS for tumor growth and survival is unequivocal. Dozens of studies show that knockdown of FAS with siRNA arrests the tumor cell cycle at G1/S and causes tumor cell apoptosis. We will not review the literature on the requirement for FAS here because much of it is directly supported by our prior publications [18,5,19-24]. Also, more than two dozen review articles have been written on this topic where strong arguments are made for FAS as a drug target (a good sampling of these reviews [25-29]). Several studies, including one by Prof. Smith, the assay provider [30], show that inhibitors of FAS may also work well to sensitize tumor cells to other widely used anti-cancer drugs including paclitaxel and Herceptin [30-36]There are two primary uses for the probe described here; i) to assist in the study of FAS in cell-based studies, and ii) to serve as the foundation for further medicinal chemistry efforts to develop a drug that selectively targets human FAS.
FAS condenses acetyl CoA with malonyl CoA In the first step in its reaction cycle. Subsequent rounds of condensation of acetyl CoA produce palmitate, which remains bound to the acyl carrier domain of FAS. The thioesterase domain, the target of the probe described here, cleaves the thioester bond with the acyl carrier protein, liberating palmitate from the enzyme. By blocking the active site of the FAS thioesterase, the probe prevents cleavage of palmitate from the acyl carrier protein, and halts palmitate biosynthesis.

Figure 1Schematic diagram of FAS showing enzymatic steps involved in fatty acid biosynthesis leading to palmitic acid
FAS is comprised of six enzymatic domains and an acyl-carrier protein (ACP) that work together to perform the following chemical steps leading to synthesis of palmitate: The malonyl/acetyl transferase domain (1) transfers an acetyl group onto the ACP. It is then translocated to the active-site cysteine by α-ketoacyl synthase (2). This position, marked “R”, also serves as the loading position for the growing acyl chain in subsequent iterations. The malonyl/acetyl transferase domain (1) then transfers a malonyl group to the ACP, and the two are condensed (2) into a four-carbon product bound to the enzyme through the thiol of the ACP. The ketoacyl reductase (3) reduces the ketone at C-3 to an alcohol. The dehydrase (4) further reduces the alcohol to an alkene. The enoyl reductase domain (5) further reduces the alkene bond to an alkane, and the ACP-bound chain is translocated back to the active-site cysteine (2). Steps 2-5 are then repeated six times to yield a 16-carbon, fully saturated palmitic acid bound to the ACP. The palmitate is released from FAS by the enzyme's intrinsic thioesterase domain (green arrow), which is the target of this study.
Several key questions on the function of FAS can be addressed by using ML356 in cell-based studies. First, there is no information on how inhibition of FAS might impacts flux through other branches of central carbon metabolism (i.e. glycolysis, TCA cycle, glutaminolysis etc.). It is reasonable to hypothesize that a FAS blockade will increase the levels of the two key substrates, acetyl Co A and malonyl CoA. The accumulation of these metabolites is likely to feedback into glycolysis and/or the TCA cycle. Levels of the NADPH cofactor are also likely to increase, perhaps inducing a cellular redox stress. ML356 makes it possible to study these issues.
In addition, there is not a good understanding of how inhibition of FAS elicits apoptosis in tumor cells. While we know that FAS blockade induces cell death in breast cancer cells through the extraneous apoptotic pathway (caspase 8), it is not at all clear why inhibition of FAS causes this effect. It may relate to either lack of cellular palmitate, or redox stress mentioned previously. ML356 makes it possible to study these issues.
There are also studies indicating the inhibition of FAS interferes with signaling through KDR, the VEGF receptor, and thereby inhibits angiogenesis [37]. However, the mechanism of inactivation of KDR has not been studied in any detail. The pharmacologic approach is especially important in studying angiogenesis because siRNA and shRNA methods are fraught with difficulty in primary cultures of endothelial cells, and are extremely time-consuming and expensive in animal models. ML356 makes it possible to understand the molecular detail connecting inhibition of FAS with KDR and VEGF signaling.
There is now widespread interest in developing precision (or personalized) medicine, especially for oncology. Gene expression and deep sequencing studies have now identified tumor cell lines that are correlates of the major sub-types of most human cancers. ML356 can be used to determine which tumor subtypes and sensitive to a FAS blockade, and to correlate this sensitivity with specific gene expression, or even genomic signatures. Such studies will go a long way toward understanding the genetic basis of the FAS-dependent phenotype in tumors.
ML356 could also be useful for screening for compounds (even existing drugs) that synergize (or are additive) with FAS blockade in halting growth and killing tumor cells, and/or endothelial cells.
ML356 can also serve as the lead structure for further medicinal chemistry to create additional probes to study the function of FAS in vivo. To our knowledge, no thioesterase (TE) has ever been targeted for drug development. Inhibitors/probes against FAS will be of value for research purposes in the development of single agent therapy against tumors, potentially as chemo-sensitizers. They may also show promise as anti-angiogenic and anti-obesity agents. Follow on derivatives of ML356 with good bioavailablity and pharmacokinetic properties will facilitate the first rigorous test of the idea that fatty acid synthase is necessary for tumor onset and/or progression in both xenograft, and genetic models of cancer. Similar studies can be done in animal models of angiogenesis, and obesity.
2. Materials and Methods
2.1. Assays
The detailed description and protocols for the primary HTS and additional assays can be found in the “Assay Description” section in the PubChem BioAssay (http://pubchem.ncbi.nlm.nih.gov/) view under the AIDs as listed in Table 2.
Table 2
Summary of Assays and AIDs.
2.2. Probe Chemical Characterization
Chemical name of probe compound. The IUPAC name of the probe [ML356] is 2-ethyl-N-(4-(4-(morpholinosulfonyl)phenyl)thiazol-2-yl)butanamide (PubChem currently does not have an IUPAC name). The actual batch prepared, tested and submitted to the MLSMR is SID 160658425 corresponding to CID 70680643.

Figure 2Structure of ML356
Synthesis and Structural Verification of probe SID 160658425 corresponding to CID 70680643 (ML356) – See Scheme 1
%3B%20b.%20Bromine%2C%20HBr%2045%25%20solution%2C%20chloroform%2C%200%20%B0C%20to%20RT%2C%20overnight%20(60%25)%3B%20c.%20Thiourea%2C%20NaHCO3%2C%20THF%2C%20RT%2C%20overnight%20(83%25)%3B%20d.%202-Ethylbutyoric%20chloride%2C%20dry%20pyridine%2C%20dry%20dichloromethane%2C%2060%B0C%20reflux%2C%20overnight%20(65%25).&p=BOOKS&id=189923_ml356f4.jpg "Click on image to zoom")
Scheme 1Synthesis of ML356, conditions: a. Morpholine, DIPEA, DCM, RT, overnight (78%); b. Bromine, HBr 45% solution, chloroform, 0 °C to RT, overnight (60%); c. Thiourea, NaHCO3, THF, RT, overnight (83%); d. 2-Ethylbutyoric chloride, dry pyridine, dry dichloromethane, 60°C reflux, overnight (65%)
Figure 31H NMR and LC-MS spectra of ML356
.&p=BOOKS&id=189923_ml356f5.jpg "Click on image to zoom")
Figure 3A1H NMR Spectrum of ML356 (500 MHz, Methanol-d4)
.&p=BOOKS&id=189923_ml356f6.jpg "Click on image to zoom")
Figure 3B13C NMR Spectrum of ML356 (125 MHz, Methanol-d4)
.&p=BOOKS&id=189923_ml356f7.jpg "Click on image to zoom")
Figure 3CLC/MS for ML356 and MS scan (inset)
Solubility and Stability of probe ML356 in PBS at room temperature: The solubility of ML356 was investigated in aqueous buffers at room temperature (see Figure 4). As noted in the Summary of in vitro ADME/T properties (see Table 4), ML356 has a poor solubility in aqueous buffers. However, ML356 appears very chemically stable in both PBS and 1:1 PBS:acetonitrile,

Figure 4Stability of ML356 in 1× PBS and 50:50 PBS:ACN
Table 4Summary of in vitro ADME Properties of FAS-TE inhibitor probe ML356
| Aqueous Solubility in pION's buffer (μg/mL) [μM]a pH 5.0/6.2/7.4 | 0.19/0.06/0.13 [0.45 /0.14 /0.31] | |
| Aqueous Solubility in 1× PBS, pH 7.4 (μg/mL) [μM]a | 0.02 [0.05] | |
| Chemical Stability in 1× PBS pH 7.4 / with 50% ACN (% remaining after 48hrs) | 100/100 | |
| PAMPA Permeability, Pe (×10-6 cm/s) Donor pH: 5.0 / 6.2 / 7.4 Acceptor pH: 7.4 | 914/961/653 | |
| Plasma Protein Binding (% Bound) | Human 1 μM / 10 μM | 96.60/97.47 |
| Mouse 1 μM / 10 μM | 96.19/96.63 | |
| Plasma Stability (% Remaining at 3 hrs) Human/Mouse | 100/100 | |
| Hepatic Microsome Stability (% Remaining at 1hr) Human/Mouse | 4.93/6.37 | |
| Toxicity Towards Fa2N-4 Immortalized Human Hepatocytes LC50 (μM) | >50 | |
- a
Solubility also expressed in molar units (μM) as indicated in italicized [bracketed values], in addition to more traditional μg/mL units.
MLS# verifying submission of probe molecule and five related samples submitted to the SMR collection. This probe is not commercially available. Samples of the probe ML356 (>25 mg), and five analogs of each (>20 mg), synthesized at SBCCG were submitted to MLSMR (Table 3, > 25 mg will be provided to the Assay Provider (Dr. Jeffery Smith) and >50 mg of the probe will be retained by SBCCG for future requests.
Table 3
Probe and Analog Submissions to MLSMR (BioFocus DPI) for FAS Inhibitors ML356.
2.3. Probe Preparation
Synthesis of probe ML356 was readily prepared in 4 steps from commercially available starting materials. (Compounds are numbers as in Scheme 1)
Preparation of 1-(4-(morpholinosulfonyl)phenyl)ethanone [2]
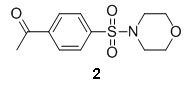
4-Acetylbenzenesufonyl chloride 1 (3.5 g, 16 mmol) and 3 ml of DIPEA were dissolved in 100 ml of dichloromethane. To the resulting mixture was added morpholine (4.0 mL, 46 mmol) and the reaction was stirred at room temperature overnight. When the reaction was determined to be complete by HPLC, the reaction mixture was concentrated under reduced pressure. The resulting oil was chromatographed on silica gel and eluted with ethyl acetate and dichoromethane (0:100 to 30:70 gradient) to yield 3.4 g of product 2 (78 % yield). MS (EI) m/z 270 (M+1).
Preparation of 2-bromo-1-(4-(morpholinosulfonyl)phenyl)ethanone [3]
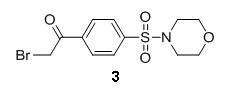
1-(4-(morpholinosulfonyl)phenyl)ethanone 2 (3.4 g, 12.6 mmol) and 3 drops of HBr 45% solution were dissolved in 100 ml of chloroform in an ice-water bath. To the resulting mixture was added dropwise bromine (0.64 ml, 12.4 mmol) in 5 ml of chloroform and the reaction was stirred overnight, gradually warming to room temperature. When the reaction was determined to be complete by HPLC, the reaction mixture was washed with a sat. sodium thiosulfate solution and separated. The organic layer was dried and concentrated under reduced pressure. The resulting oil was chromatographed on silica gel and eluted with ethyl acetate and dichoromethane (0:100 to 30:70 gradient) to yield 2.6 g of product 3 (60 % yield). MS (EI) m/z 350 (M+1). 1H NMR (400 MHz, CDCl3) δ(ppm) 3.04 (m, 4H), 3.75 (m, 4H), 4.45 (m, 2H), 7.87 (d, 2H), 8.15 (d, 2H).
Preparation of 4-(4-(morpholinosulfonyl)phenyl)thiazol-2-amine [2]

2-bromo-1-(4-(morpholinosulfonyl)phenyl)ethanone 3 (2.6 g, 7.4 mmol) and sodium bicarbonate (1.3 g, 16 mmol) were mixed in 100 ml of dry THF. To the resulting mixture was added thiourea (1.0 g, 13 mmol) and the reaction was stirred at room temperature overnight. When the reaction was determined to be complete by HPLC, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in dichloromethane and washed with water. The organic layer was dried and concentrated and was chromatographed on silica gel and eluted with ethyl acetate and dichoromethane (0:100 to 40:60 gradient) to yield 2.0 g of product 4 (83 % yield). 1H NMR (400 MHz, DMSO-d6) δ(ppm) 2.86 (m, 4H), 3.62 (m, 4H), 7.18 (s, 2H), 7.30 (s, 1H), 7.70 (d, 2H), 8.03 (d, 2H). 13C NMR (400 MHz, DMSO-d6) δ(ppm) 39.9, 65.2, 105.0, 126.1, 128.1, 132.4, 139.2, 148.1, 168.5. MS (EI) m/z 326 (M+1).
Preparation of 2-ethyl-N-(4-(4-(morpholinosulfonyl)phenyl)thiazol-2-yl)butanamide [3]

4-(4-(morpholinosulfonyl)phenyl)thiazol-2-amine 4 (0.5 g, 1.5 mmol) and 1.0 mL of dry pyridine were dissolved in dry dichoromethane (50 mL). To the resulting mixture was added 2-ethylbutyryl chloride (0.5 mL, 3.7 mmol) and the reaction was refluxed at 60 °C overnight. When the reaction was determined to be complete by HPLC, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting oil was chromatographed on silica gel and eluted with ethyl acetate and dichoromethane (0:100 to 20:80 gradient) to yield 0.42 g of product 5 (65 % yield). 1H NMR (400 MHz, CDCl3) δ(ppm) 0.91 (m, 6H), 1.61 (m, 2H), 1.71 (m, 2H), 2.20 (m, 1H), 3.04 (m, 4H), 3.75 (m, 4H), 7.33 (s, 1H), 7.85 (d, 2H), 8.02 (d, 2H), 9.57 (s, 1H). 13C NMR (400 MHz, CDCl3) δ(ppm) 11.9, 25.5, 46.0, 50.9, 66.1, 110.6, 126.5, 128.4, 133.9, 138.8, 147.7, 158.3, 174.2. MS (EI) m/z 424 (M+1).
3. Results
3.1. Dose Response Curves for Probe
The in vitro biochemical potency (IC50) ML356 against FAS-TE is 334 nM (Figure 5).
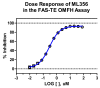
Figure 5
Potency of ML356 against the thioester domain of fatty acid synthase (FAS-TE).
3.5. Cellular Activity
The ability of probes to block biosynthesis of palmitate in whole cells (prostate cancer PC-3 cells) was measured by feeding cells isotopically labeled glucose (13C-glucose) and measuring the incorporation of 13C into palmitate by GC-MS. These assays indicate the inhibitory activity of the probe ML356 and active analog in whole cells (Figure 6), whereas inactive analog (MLS-0472100) did not inhibit palmitate biosynthesis.

3.3. Profiling Assays
ML356 was evaluated in a detailed in vitro pharmacology screen as shown in Table 4:
ML356 has poor solubility in aqueous media.
The PAMPA (Parallel Artificial Membrane Permeability Assay) assay is used as an in vitro model of passive, transcellular permeability. An artificial membrane immobilized on a filter is placed between a donor and acceptor compartment. At the start of the test, drug is introduced in the donor compartment. Following the permeation period, the concentration of drug in the donor and acceptor compartments is measured using UV spectroscopy. ML356 exhibited very good permeability across a range of pH of the donor compartment.
Plasma protein binding is a measure of a drug's efficiency to bind to the proteins within blood plasma. The less bound a drug is, the more efficiently it can traverse cell membranes or diffuse. Highly plasma protein bound drugs are confined to the vascular space, thereby having a relatively low volume of distribution. In contrast, drugs that remain largely unbound in plasma are generally available for distribution to other organs and tissues. ML356 was highly plasma protein bound.
Plasma stability is a measure of the stability of small molecules and peptides in plasma and is an important parameter, which can strongly influence the in vivo efficacy of a test compound. Drug candidates are exposed to enzymatic processes (proteinases, esterases) in plasma, and they can undergo intramolecular rearrangement or bind irreversibly (covalently) to proteins. ML356 showed good stability in both human plasma and mouse plasma.
The microsomal stability assay is commonly used to rank compounds according to their metabolic stability. This assay addresses the pharmacologic question of how long the parent compound will remain circulating in plasma within the body. ML356 showed poor stability in human and mouse liver microsomes.
ML356 showed no toxicity (>50 μM) towards immortalized Fa2-N4 human hepatocytes.
4. Discussion
The ultimate objective of this proposal is to develop drug-like inhibitors of the thioesterase (TE) domain of fatty acid synthase (FAS: NP004095.4, GI:41872631), an enzyme that is essential for growth of solid tumors. We hypothesize that such compounds will block tumor growth in vivo because FAS is up-regulated in all the major solid tumors [18], and in most cases its expression is indicative of poor prognosis [1,2,11]. Knockdown of FAS with siRNA halts tumor cell proliferation and selectively induces apoptosis of tumor cells [30,33]. The correlation between expression of FAS and poor prognosis strongly suggests that this enzyme is mechanistically linked to disease progression, providing a strong rationale for developing inhibitors of FAS. The overarching objective of this study is to identify novel inhibitors of the thioesterase domain of FAS that are potent, selective, and stable.
To our knowledge, no thioesterase (TE) has ever been targeted for drug development. Consequently, the unique enzymatic target and the new chemical entities that will be developed point to the novelty of the study. The work proposed here is likely to pave the way for discovery of probes and drugs to target thioesterases similar to those involved in polyketide synthesis, mycolic acid biosynthesis from Mycobacterium tuberculosis, siderophore biosynthesis in bacteria, etc. This illustrates the metabolic context of the FAS enzyme and highlights the rationale for the druggabiiity of this target and selectivity against other off-target enzymes.
4.1. Comparison to Existing Art and How the New Probe is an Improvement
Several inhibitors of human FAS have been reported, but almost all of these have serious limitations including, complex synthetic procedures, poor solubility, lack of potency, covalent modes of binding, and perhaps most importantly, lack of selectivity (for review see [27]). The three most commonly used FAS antagonists are cerulenin (or its derivative C75), and orlistat.
Cerulenin a fatty-acid like dodecadienamide is natural product with antibiotic properties, that was found to inhibit human FAS and to be cytotoxic to tumor cells [2]. However, cerulenin is an irreversible inhibitor (epoxide) of FAS β-ketoacyl synthase, and is not selective for FAS; it affects many other processes (protein acylation, cholesterol synthesis and proteolysis). C75 is a synthetic analog of cerulenin that blocks tumor-cell proliferation and induces apoptosis, has anti-tumor activity in mouse models of androgen-independent prostate cancer [38], breast cancer [3], mesothelioma [39], and ovarian cancer [12]. But C75 is a weakly potent (mM-range) inhibitor of FAS β-ketoacyl synthase, acid still contains obviously reactive moieties (lactone and electrophilic exo double bond). C75 also lacks selectivity for FAS; it is a potent agonist of carnitine palmitoyl transferase 1 (CPT1), and through this action it simulates fatty acid oxidation and the production of ATP [40], and causes weight loss in animals. So C75 lacks selectivity, and has other known targets, making it impossible to use this compound to draw conclusions on FAS.
The assay provider's group (Dr. Smith) has published several papers on the use of orlistat as a FAS inhibitor [41,42]. Importantly, orlistat is not specific to FAS; its anti-obesity effect has been associated with irreversible inactivation of gastric and pancreatic lipases. Although orlistat is an interesting lead antagonist of FAS, it contains a reactive β-lactone that is not optimal for drug/probe development. The reactive β-lactone leads to dead end inhibition of FAS, so once the drug is on board, removal of the agent is dependent upon the half-life of FAS; halting administration of the drug is of little value if any acute toxicity were dose-limiting. Furthermore, the reactive group is likely to react with other proteins/enzymes, as well as with plasma and tissue constituents, leading to a complicated pharmacokinetic profile. Finally, orlistat has poor solubility (cLogP = ∼14), making it almost impossible to determine the precise dose of the compounds that inhibits FAS in cells or in animals. Dr. Smiths group, along with Dan Romo's group at Texas A&M attempted to generate more soluble and selective orlistat-derivatives, but complex synthetic procedures hindered this effort [41-44].
The green tea polyphenol epigallocatechin-3-gallate (EGCG) and other naturally occurring flavonoids (i.e., luteolin, quercetin, and kaempferol), as well as the antibiotic triclosan, are reported to inhibit FAS and induce apoptotic cell death [28]. Another polyphenol/polycatechol compound G284CM has been reported to inhibit FAS; however, its exact structure is not available, polyphenols are prone to oxidation and oligomerization, and no proof of its direct interaction with FAS has been disclosed. Irrespective of G284CM primary target, chemical liabilities affiliated with polyphenol/catechol scaffold, makes it un-drug-like. Orlistat, an antiobesity drug, irreversibly modifies FAS TE domain active site. However, it is not specific to FAS; its antiobesity effect has been associated with irreversible inactivation of gastric and pancreatic lipases.
In summary, all these compounds, while known to inhibit FAS irreversibly, are certainly not selective for FAS as they all have other reported targets consistent with the non-specificity of alkylation prone reactive moieties. Finally, there is no known reversible inhibitor of thioesterase domain of FAS to date.
A recent SciFinder search conducted by Dr. Anthony Pinkerton on November 27, 2011, found an additional recent report of a FAS inhibitor, the antibiotic platensimycin (Figure 7). Platensimycin has been shown to concentrate in the liver after oral administration to mice, and potently inhibits hepatic de novo lipogenesis, reduces fatty acid oxidation, and increases glucose oxidation in mouse models of diabetes [45]. It, however, is a natural product of high molecular weight (m.w. 441) and complex structure originally isolated as broad spectrum Gram-positive antibiotic produced and isolated from Streptomyces platensis [46]. Synthesis yields a racemic mixture, therefore, simpler chemically tractable probes are still needed. Nor is there any evidence of its selectivity for FAS over other human thioesterases.
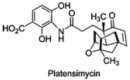
Figure 7
Structure of Platensimycin.
A more recent SciFinder search conducted by Dr. Robert J. Ardecky on April 1, 2013, found one new reference that describes compounds that inhibit the recombinant domain of FASN. Some compounds show the ability to inhibit FASN in living cells and some compounds showed the ability to inhibit in vitro cancer cell growth. The general structure of these compounds is illustrated in Figure 8: The IC50 values of the best compounds were in 2.35 μM to 10 μM range. (PCT Int. Appl. (2012), WO 2012064632 A1 20120518)
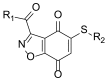
Figure 8
Markush structure from PCT Int. Appl. (2012), WO 2012064632 A1 20120518).
In this report we have identified the most potent and first-in-class FAS TE Inhibitor known, ML356 (CID 70680643). More importantly, ML356, is active in a cellular based assay and has the ability to block cellular fatty acid biosynthesis. ML356 is chemically stable and will serve as a unique biochemical probe.
This compound will be particularly valuable to the cancer biology, obesity research, and metabolism communities, as the biochemical and cellular processes that control FAS can now be further explored by pharmacologic inhibition. The original goal for this project was to screen for reversible antagonists of the thioesterase domain of FAS that will prevent cellular fatty acid biosynthesis, selectively induce tumor cell apoptosis, and be useful in treating tumors. We have identified a compound, ML356 that could potentially meet our goals.
Evidence of selectivity for FAS over other human thioesterases
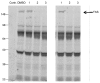
Studies were conducted to determine the selectivity of ML356 for FAS over other human thioesterases. Thioesterases are members of the serine hydrolase family, which also includes a number of other types of enzymes including proteases, dipeptidyl peptidases, epoxide hydrolases etc. To help assess the selectivity of ML356 we used in an activity-based probe polyethyleneglycol-6-carboxytetra-methyl-rhodamine (FP-PEG-TAMRA) that binds covalently to the catalytic serine of all serine hydrolases [47]. These studies were done by assessing the ability of ML356 to compete for the binding of the activity-based probe in whole cell lysates. Several analogs of probe ML356 were tested in this assay using lysates of the prostate tumor line (PC3), which expresses FAS, along with numerous other serine hydrolases [48]. The PC-3 lysate was incubated with 8 different FAS inhibitors from the screen, including ML356 (Figure 9, lane 3 both gels). Then the activity-based probe (FP-PEG-TAMRA) was added to each sample to label the serine hydrolases in the lysate. Samples were resolved by 10% SDS-PAGE and activity-based labeling was visualized at 535 nm. The probe ML356 (Figure 9, #3:CID1811788, MLS-0472190) and an active analog (Figure 9, #2: MLS-0380827) showed inhibition of FAS, whereas an inactive control compound (Figure 9, #1: MLS-0472100) did not. These results show that the probe ML356 and key analogs from the screen block the binding of FP-TAMRA to FAS. However, there is no competition for activity based labeling against any of the other serine hydrolases in the lysate. These findings indicate that ML356 is highly selective for the thioesterase domain of FAS.
Additional, studies were done to test the selectivity of ML356 for the FAS thioesterase over its most closely related human homolog and enzyme called ACOT4 (Figure 10). Recombinant forms of each enzyme were incubated with ML356 [CID 70680643], and some other analogues of this compound. FP-TAMRA was included in this mixture to label the active site of the FAS thioesterase and ACOT4. ML356 and its active analogs blocked the binding of FP-TAMRA to the active site of the FAS thioesterase but were without effect on activity labeling of ACOT4 (Figure 10A). These findings further attest to the high degree of selectivity of ML356 for the FAS thioesterase.
At this juncture the probes have most of the biochemical properties that we set out to identify; reasonable potency against the recombinant thioesterase, the ability to block the active site of the thioesterase in the context of the FAS holoenzyme, the ability to block fatty acid biosynthesis in whole cells, and selectivity against the most closely related human thioesterase, ACOT4. Together these properties provide the rationale for engaging in a medicinal chemistry effort to optimize potency, retain selectivity, and to improve the pharmacologic properties of the compound so that it has a pharmacokinetic profile suitable for use in long-term animal studies. This medicinal chemistry effort has already begun in the laboratories of the PI and Co-PI. Our objective is to use the optimized probes to study tumor metabolism and tumor growth.
5. References
- 1.
- Shurbaji MS, Kuhajda FP, Pasternack GR, Thurmond TS. Expression of oncogenic antigen 519 (OA-519) in prostate cancer is a potential prognostic indicator. Am J Clin Pathol. 1992;97(5):686–691. [PubMed: 1374214]
- 2.
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A. 1994;91(14):6379–6383. [PMC free article: PMC44205] [PubMed: 8022791]
- 3.
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Research. 2000;60(2):213–218. [PubMed: 10667561]
- 4.
- Myers RB, Oelschlager DK, Weiss HL, Frost AR, Grizzle WE. Fatty acid synthase: an early molecular marker of progression of prostatic adenocarcinoma to androgen independence. J Urol. 2001;165(3):1027–1032. [PubMed: 11176534]
- 5.
- Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen JV. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol Endocrinol. 2001;15(10):1817–1828. [PubMed: 11579213]
- 6.
- Bull JH, Ellison G, Patel A, Muir G, Walker M, Underwood M, Khan F, Paskins L. Identification of potential diagnostic markers of prostate cancer and prostatic intraepithelial neoplasia using cDNA microarray. Br J Cancer. 2001;84(11):1512–1519. [PMC free article: PMC2363654] [PubMed: 11384102]
- 7.
- Alo PL, Visca P, Trombetta G, Mangoni A, Lenti L, Monaco S, Botti C, Serpieri DE, Di Tondo U. Fatty acid synthase (FAS) predictive strength in poorly differentiated early breast carcinomas. Tumori. 1999;85(1):35–40. [PubMed: 10228495]
- 8.
- Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77(3):474–482. [PubMed: 8630954]
- 9.
- Visca P, Alo PL, Del Nonno F, Botti C, Trombetta G, Marandino F, Filippi S, Di Tondo U, Donnorso RP. Immunohistochemical expression of fatty acid synthase, apoptotic-regulating genes, proliferating factors, and ras protein product in colorectal adenomas, carcinomas, and adjacent nonneoplastic mucosa. Clinical Cancer Research. 1999;5(12):4111–4118. [PubMed: 10632348]
- 10.
- Rashid A, Pizer ES, Moga M, Milgraum LZ, Zahurak M, Pasternack GR, Kuhajda FP, Hamilton SR. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. American Journal of Pathology. 1997;150(1):201–208. [PMC free article: PMC1858511] [PubMed: 9006336]
- 11.
- Gansler TS, Hardman W III, Hunt DA, Schaffel S, Hennigar RA. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum Pathol. 1997;28(6):686–692. [PubMed: 9191002]
- 12.
- Pizer ES, Jackisch C, Wood FD, Pasternack GR, Davidson NE, Kuhajda FP. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996;56(12):2745–2747. [PubMed: 8665507]
- 13.
- Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. 1996;27(9):917–921. doi: S0046-8177(96)90218-X. [PubMed: 8816886]
- 14.
- Ogino S, Nosho K, Meyerhardt JA, Kirkner GJ, Chan AT, Kawasaki T, Giovannucci EL, Loda M, Fuchs CS. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J Clin Oncol. 2008;26(35):5713–5720. JCO.2008.18.2675. [PMC free article: PMC2630484] [PubMed: 18955444] [CrossRef]
- 15.
- Camassei FD, Jenkner A, Rava L, Bosman C, Francalanci P, Donfrancesco A, Alo PL, Boldrini R. Expression of the lipogenic enzyme fatty acid synthase (FAS) as a predictor of poor outcome in nephroblastoma: An interinstitutional study. Med Pediatr Oncol. 2003;40(5):302–308. [PubMed: 12652618]
- 16.
- Horiguchi A, Asano T, Ito K, Sumitomo M, Hayakawa M. Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J Urol. 2008;180(3):1137–1140. S0022-5347(08)01215-9. [PubMed: 18639282] [CrossRef]
- 17.
- Silva SD, Perez DE, Nishimoto IN, Alves FA, Pinto CA, Kowalski LP, Graner E. Fatty acid synthase expression in squamous cell carcinoma of the tongue: clinicopathological findings. Oral Dis. 2008;14(4):376–382. ODI1395. [PubMed: 18410580] [CrossRef]
- 18.
- Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A. 2000;97(7):3450–3454. [PMC free article: PMC16260] [PubMed: 10716717]
- 19.
- Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19(1-2):139–145. [PubMed: 11191053]
- 20.
- Pages G, Milanini J, Richard DE, Berra E, Gothie E, Vinals F, Pouyssegur J. Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann N Y Acad Sci. 2000;902:187–200. [PubMed: 10865838]
- 21.
- Pages G, Berra E, Milanini J, Levy AP, Pouyssegur J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J Biol Chem. 2000;275(34):26484–26491. [PubMed: 10849421]
- 22.
- Cao Y. Molecular mechanisms and therapeutic development of angiogenesis inhibitors. Adv Cancer Res. 2008;100:113–131. [PubMed: 18620094]
- 23.
- Chung CC, Ohwaki K, Schneeweis JE, Stec E, Varnerin JP, Goudreau PN, Chang A, Cassaday J, Yang L, Yamakawa T, Kornienko O, Hodder P, Inglese J, Ferrer M, Strulovici B, Kusunoki J, Tota MR, Takagi T. A fluorescence-based thiol quantification assay for ultra-high-throughput screening for inhibitors of coenzyme A production. Assay Drug Dev Technol. 2008;6(3):361–374. [PubMed: 18452391]
- 24.
- Elice F, Jacoub J, Rickles FR, Falanga A, Rodeghiero F. Hemostatic complications of angiogenesis inhibitors in cancer patients. Am J Hematol. 2008 [PubMed: 18819092]
- 25.
- Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100(9):1369–1372. 6605007. [PMC free article: PMC2694429] [PubMed: 19352381] [CrossRef]
- 26.
- Young CD, Anderson SM. Sugar and fat - that's where it's at: metabolic changes in tumors. Breast Cancer Res. 2008;10(1):202. bcr1852. [PMC free article: PMC2374962] [PubMed: 18304378] [CrossRef]
- 27.
- Kridel SJ, Lowther WT, Pemble CWt. Fatty acid synthase inhibitors: new directions for oncology. Expert Opin Investig Drugs. 2007;16(11):1817–1829. [PubMed: 17970640] [CrossRef]
- 28.
- Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Curr Pharm Biotechnol. 2006;7(6):483–493. [PubMed: 17168665]
- 29.
- Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66(12):5977–5980. 66/12/5977. [PubMed: 16778164] [CrossRef]
- 30.
- Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty acid synthase induces caspase 8-mediated tumor cell apoptosis by Up-regulating DDIT4. J Biol Chem. 2008 [PMC free article: PMC2581575] [PubMed: 18796435]
- 31.
- Carlo-Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res. 2007;13(8):2313–2317. [PubMed: 17438088]
- 32.
- Menendez JA, Vellon L, Lupu R. Antitumoral actions of the anti-obesity drug orlistat (XenicalTM) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene. Ann Oncol. 2005;16(8):1253–1267. mdi239. [PubMed: 15870086] [CrossRef]
- 33.
- Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A. 2004;101(29):10715–10720. 0403390101. [PMC free article: PMC490000] [PubMed: 15235125] [CrossRef]
- 34.
- Menendez JA, Colomer R, Lupu R. Inhibition of tumor-associated fatty acid synthase activity enhances vinorelbine (Navelbine)-induced cytotoxicity and apoptotic cell death in human breast cancer cells. Oncol Rep. 2004;12(2):411–422. [PubMed: 15254710]
- 35.
- Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008;41(1):59–85. CPR498. [PMC free article: PMC6496011] [PubMed: 18211286] [CrossRef]
- 36.
- Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007;31(4):769–776. [PubMed: 17786307]
- 37.
- Browne CD, Hindmarsh EJ, Smith JW. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J. 2006;20(12):2027–2035. [PubMed: 17012255] [CrossRef]
- 38.
- Pizer ES, Pflug BR, Bova GS, Han WF, Udan MS, Nelson JB. Increased fatty acid synthase as a therapeutic target in androgen-independent prostate cancer progression. Prostate. 2001;47(2):102–110. [PubMed: 11340632]
- 39.
- Gabrielson EW, Pinn ML, Testa JR, Kuhajda FP. Increased fatty acid synthase is a therapeutic target in mesothelioma. Clinical Cancer Research. 2001;7(1):153–157. [PubMed: 11205903]
- 40.
- Thupari JN, Landree LE, Ronnett GV, Kuhajda FP. C75 increases peripheral energy utilization and fatty acid oxidation in diet-induced obesity. Proc Natl Acad Sci U S A. 2002;99(14):9498–9502. 132128899. [PMC free article: PMC123169] [PubMed: 12060712] [CrossRef]
- 41.
- Richardson AD, Y C, Osterman A, Smith JW. Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res Treat. 2008;110(2):297–307. [PMC free article: PMC2440942] [PubMed: 17879159] [CrossRef]
- 42.
- Zhang W, Richardson RD, Chamni S, Smith JW, Romo D. Beta-lactam congeners of orlistat as inhibitors of fatty acid synthase. Bioorg Med Chem Lett. 2008;18(7):2491–2494. [PubMed: 18343106] [CrossRef]
- 43.
- Rivkin A, Kim YR, Goulet MT, Bays N, Hill AD, Kariv I, Krauss S, Ginanni N, Strack PR, Kohl NE, Chung CC, Varnerin JP, Goudreau PN, Chang A, Tota MR, Munoz B. 3-Aryl-4-hydroxyquinolin-2(1H)-one derivatives as type I fatty acid synthase inhibitors. Bioorg Med Chem Lett. 2006;16(17):4620–4623. [PubMed: 16784844] [CrossRef]
- 44.
- Purohit VC, Richardson RD, Smith JW, Romo D. Practical, catalytic, asymmetric synthesis of beta-lactones via a sequential ketene dimerization/hydrogenation process: inhibitors of the thioesterase domain of fatty acid synthase. J Org Chem. 2006;71(12):4549–4558. [PubMed: 16749788] [CrossRef]
- 45.
- Wu M, Singh SB, Wang J, Chung CC, Salituro G, Karanam BV, Lee SH, Powles M, Ellsworth KP, Lassman ME, Miller C, Myers RW, Tota MR, Zhang BB, Li C. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc Natl Acad Sci U S A. 2011;108(13):5378–5383. [PMC free article: PMC3069196] [PubMed: 21389266] [CrossRef]
- 46.
- Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. Isolation, structure, and absolute stereochemistry of platensimycin, a broad spectrum antibiotic discovered using an antisense differential sensitivity strategy. J Am Chem Soc. 2006;128(36):11916–11920. [PubMed: 16953632] [CrossRef]
- 47.
- Knowles LM, Axelrod F, Browne CD, Smith JW. A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2. J Biol Chem. 2004;279(29):30540–30545. [PubMed: 15138278] [CrossRef]
- 48.
- Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64(6):2070–2075. [PubMed: 15026345]
- 49.
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [PMC free article: PMC5554542] [PubMed: 22930834]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-kappaB.[Int J Oncol. 2004]Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-kappaB.Menendez JA, Mehmi I, Atlas E, Colomer R, Lupu R. Int J Oncol. 2004 Mar; 24(3):591-608.
- Computational screening of fatty acid synthase inhibitors against thioesterase domain.[J Biomol Struct Dyn. 2018]Computational screening of fatty acid synthase inhibitors against thioesterase domain.Panman W, Nutho B, Chamni S, Dokmaisrijan S, Kungwan N, Rungrotmongkol T. J Biomol Struct Dyn. 2018 Nov; 36(15):4114-4125. Epub 2017 Dec 7.
- Review Structure and function of animal fatty acid synthase.[Lipids. 2004]Review Structure and function of animal fatty acid synthase.Chirala SS, Wakil SJ. Lipids. 2004 Nov; 39(11):1045-53.
- Mapping of functional interactions between domains of the animal fatty acid synthase by mutant complementation in vitro.[Biochemistry. 1997]Mapping of functional interactions between domains of the animal fatty acid synthase by mutant complementation in vitro.Joshi AK, Witkowski A, Smith S. Biochemistry. 1997 Feb 25; 36(8):2316-22.
- Review Fatty acid synthase-catalyzed de novo fatty acid biosynthesis: from anabolic-energy-storage pathway in normal tissues to jack-of-all-trades in cancer cells.[Arch Immunol Ther Exp (Warsz)....]Review Fatty acid synthase-catalyzed de novo fatty acid biosynthesis: from anabolic-energy-storage pathway in normal tissues to jack-of-all-trades in cancer cells.Menendez JA, Lupu R. Arch Immunol Ther Exp (Warsz). 2004 Nov-Dec; 52(6):414-26.
- Selective inhibitors of FAS-TE - Probe Reports from the NIH Molecular Libraries ...Selective inhibitors of FAS-TE - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...