NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.
Lysosomal storage diseases (LSDs) comprise a group of at least 50 distinct genetic diseases, each one resulting from a deficiency of a particular lysosomal protein/activity or, in a few cases, from non-lysosomal activities that are involved in lysosomal biogenesis or protein maturation. Fabry disease is the second most common of the LSDs, after Gaucher disease. The reported epidemiological data are likely to be underestimates, due to missed diagnoses of these rare disorders. The positive and negative outcomes of newborn screening for Fabry disease and LSDs in general are considered. Early diagnosis and intervention before the onset of irreversible pathology will provide a substantial benefit to many of these newborns, as well as providing the opportunity for parents to receive genetic counselling. However, there can also be potential harm to the parent/newborn relationship as a consequence of knowing that the baby has an incurable disorder.
Introduction
A significant problem in gathering epidemiological data is the clinical heterogeneity present in all lysosomal storage diseases (LSDs). This leads to missed diagnoses and diagnostic confusion, which results in poor epidemiological data and an underestimation of the impact of LSDs in the community. Accurate epidemiological data are essential in order to appreciate both the specific and overall impact of LSDs on patients, families and the community, and to enable the introduction of policies that will effectively reduce this impact.
Genes and proteins
LSDs comprise a group of at least 50 distinct genetic diseases [1], each one resulting from a deficiency of a particular lysosomal protein/activity or, in a few cases, from non-lysosomal activities that are involved in lysosomal biogenesis or protein maturation. The number of recognized LSDs is increasing as new disorders are characterized biochemically and genetically. Over the past decade, a deficiency of cathepsin K has been described, which results in an LSD called pycnodysostosis [2], and several of the genes and proteins involved in the neuronal ceroid lipofuscinoses (Batten disease family) have been characterized. Infantile neuronal ceroid lipofuscinosis, also known as Santavuori disease, has been shown to result from a deficiency of palmitoyl protein thioesterase [3], and classic late-infantile neuronal ceroid lipofuscinosis (Jansky–Bielschowsky disease) has been shown to result from a deficiency of tripeptidyl peptidase I [4], both of which are lysosomal enzymes. At least eight genes are thought to be involved in this group of LSDs, but only six have been identified to date [5]. In addition, the protein deficiency leading to Danon disease was recently identified as the lysosome-associated membrane protein LAMP-2 [6]. Undoubtedly, many more proteins and genes are involved in LSDs but are yet to be characterized.
Most LSDs are inherited in an autosomal recessive manner, with the exception of Hunter syndrome or mucopolysaccharidosis type II (MPS II), which shows X-linked recessive inheritance; Danon disease, which is X-linked dominant; and Fabry disease, which, with a high proportion of affected females, should not be described as X-linked recessive.
Although each LSD results from mutations in a different gene and a consequent deficiency of enzyme activity or protein function, all LSDs share one common biochemical characteristic: they result in an accumulation of substrates within lysosomes. The particular substrates that are stored and the site(s) of storage vary. The substrate type is used to group the LSDs into broad categories, including the mucopolysaccharidoses, the lipidoses, the glycogenoses and the oligosaccharidoses [7]. Despite this categorization, many clinical similarities are observed between groups as well as within each group. Common clinical features of many LSDs include bone abnormalities, organomegaly, central nervous system dysfunction and coarse hair and facies.
Some LSDs have been classified into clinical subtypes (such as the Hurler/Scheie definitions of MPS I or the infantile-/juvenile-/adult-onset forms of Pompe disease), but it is clear that most LSDs have a broad continuum of clinical severity and age at presentation.
Incidence and prevalence
Limited numbers of studies have investigated the incidence of LSDs, defined as the total number of cases diagnosed within a certain period of time, divided by the total number of live births in the same period (Table 1). One of the main problems associated with obtaining accurate epidemiological data for these individually rare disorders is that, in most countries, there are numerous diagnostic centres, which compounds the problem of collecting and correlating diagnoses. In Australia, two diagnostic centres serve the entire country, as well as covering New Zealand and a portion of South-East Asia. The National Referral Laboratory for Lysosomal, Peroxisomal and Related Genetic Disorders at the Children, Youth and Women's Health Service, North Adelaide diagnoses approximately 80% of LSD patients in Australia, while the Division of Chemical Pathology at the Royal Brisbane Hospitals, Brisbane covers the remainder.
In a retrospective case study, Meikle et al. reported on the incidence of LSDs in Australia [8]. Over the period January 1980 to December 1996, 470 diagnoses of LSDs were made, representing 27 different disorders. Based on these figures, the incidence of LSDs ranges from 1 in 57 000 for Gaucher disease to as low as 1 in 4.2 million for sialidosis. The incidence of LSDs as a group was calculated to be 1 in 7700; if prenatal diagnoses were not considered, the incidence of LSDs was 1 in 9000 births. However, the neuronal ceroid lipofuscinoses were not included in this study. Estimates of the global incidence of neuronal ceroid lipofuscinoses as high as 1 in 12 500 have been reported [9]. Based on Australian diagnoses made during the period 1998–2003, the combined incidence of the three most prevalent forms of neuronal ceroid lipofuscinoses (infantile, late-infantile and juvenile) has been estimated at approximately 1 in 60 000 (personal communication, MJ Fietz; Children, Youth and Women's Health Service, North Adelaide, Australia). Furthermore, there are likely to be other LSDs that are yet to be clinically and biochemically described, suggesting that the combined incidence of LSDs may be as high as 1 in 5000.
Meikle et al. reported the incidence of Fabry hemizygotes as 1 in 117 000 [8]. No data on heterozygotes were obtained, but the incidence determined in hemizygotes could be extrapolated to give a combined incidence of 1 in 58 000.
The incidence of LSDs in Australia is not dissimilar to other countries. Poorthuis et al. [10] reported on the frequency of LSDs in The Netherlands based on 963 diagnosed cases over the period 1970–96. The combined birth prevalence for all LSDs, defined as the total number of diagnosed cases born within a certain period of time divided by the total number of live births in the same period, was calculated to be 1 in 7100 live births, with glycogen storage disease type II being the most prevalent at 1 in 50 000. The prevalence of Fabry disease was estimated at 1 in 476 000 in this population, lower than in the Australian population, and Gaucher disease was calculated to be 1 in 86 000, also somewhat lower than the Australian figure and representing 8% of all reported LSDs.
A report from Italy on inborn errors of metabolism gave a combined LSD incidence of 1 in 8275, with Gaucher disease at 1 in 40 247 (21% of all reported LSDs) [11]. A recent report from Portugal estimated the combined birth prevalence of all LSDs to be 1 in 4000 births, with Gaucher disease at 1 in 74 000 births (5% of all reported LSDs) [12]. Ozkara and Topcu reported the combined birth prevalence of the sphingolipidoses in Turkey at 1 in 21 500, with Gaucher disease at 1 in 185 000 and Fabry disease 1 in 6 700 000 [13]. However, their study included only children under 5 years of age with neurological symptoms, thereby excluding most patients with Fabry disease. Bähner and colleagues recently reported that the incidence of MPS in Germany (1 in 28 000) is similar to that reported in other populations; however, different frequencies of individual MPS subtypes were observed [14].
There have also been several reports on the incidence of particular disorders in specific populations: values as high as 1 in 18 500 for aspartylglucosaminuria in the Finnish population [15] and 1 in 3900 for Tay–Sachs disease in the Ashkenazi Jewish population [16] have been reported.
In the UK, the prevalence of Fabry disease was reported to be 1 in 366 000 [17]. This figure was based on notifications of patients seen in UK clinics who had low α-galactosidase activities. Many patients with Fabry disease are likely to have been omitted and therefore the prevalence figure is probably an underestimate.
High-risk populations
Although LSDs have a low incidence in most populations, there are a number of exceptions. The Ashkenazi Jewish population is at high risk for a number of LSDs, including Gaucher disease, which is thought to have an incidence as high as 1 in 855 Ashkenazi Jewish births [18], Tay–Sachs disease and Niemann–Pick disease [19]. The Finnish population has been reported to have a high incidence of aspartylglucosaminuria (1 in 18 500 births) [15] and infantile (1 in 13 000 births) and juvenile (1 in 21 000 births) neuronal ceroid lipofuscinosis [20]. Both communities represent populations that have been, and to some extent still are, genetically isolated either culturally or geographically. This has contributed to the founder effects that have led to the relatively high incidence of specific LSDs in these populations.
Interpretation of data
For diseases that are very rare, even a single missed diagnosis can make a large difference to the calculated birth prevalence. In addition, the methods used to calculate incidence and birth prevalence figures must also be considered when comparing data from separate studies. In the Australian study, the period used to determine the total number of births was the period in which the diagnoses were made; the assumption was made that late-onset patients diagnosed during the study (and born before the study period) were representative of late-onset patients born within the study period who had not yet presented clinically [8]. In the Dutch and Portuguese studies, the period used to calculate the birth prevalence was based on the age of the patient; thus, the assumption was made that all patients born prior to the last patient in the subgroup were identified [10, 12]. There are limitations to both methods and it is likely that ascertainment of the late-onset LSD patients was incomplete, leading to underestimation of incidence and birth prevalence. In Australia, the incidence of Pompe disease was estimated to be approximately 1 in 146 000. However, studies based on carrier detection in normal populations have indicated that the incidence may be as high as 1 in 40 000 births in both the USA [21] and The Netherlands [22]. On the basis of their findings, Martiniuk and colleagues [21] predict that many mild adult cases remain undiagnosed.
Burden of illness
The burden of LSDs on the individual and family is undoubtedly enormous, although there is relatively little documentation in this area. The cost to the community in terms of medical treatment and support is also poorly defined. Therapy is likely to be available for several LSDs in the near future. In addition, a number of technologies for newborn screening have been proposed. If the current cost of these disorders to public health systems is to be assessed accurately, thereby allowing the cost-effectiveness of therapy and/or newborn screening to be investigated, accurate prevalence values will be required. The introduction of newborn screening for LSDs will provide such accurate prevalence data.
Fabry disease
Fabry disease has been reported to be the second most common LSD, after Gaucher disease, although reported incidence and birth prevalence figures vary considerably. These figures are likely to be underestimated due to missed diagnoses, because the disease presentation can be non-specific and signs and symptoms are often mistakenly attributed to other disorders.
Renal insufficiency is a key clinical feature of Fabry disease, and it is believed that the birth prevalence and incidence of Fabry disease in patients with end-stage renal failure is underestimated [23]. In a retrospective study of 105 male patients with Fabry disease, of whom 94 were Caucasian, 74% had renal disease [24]. Linthorst et al. [23] estimated the prevalence of Fabry disease in patients on dialysis at 0.22%. Consequently, screening for Fabry disease in large renal dialysis clinics has been suggested. In France, 106 patients on haemodialysis were screened for α-galactosidase activity. One patient was identified with Fabry disease and a further seven family members of this index case carried the same mutation [25]. A study in Japan reported that 1.2% of males receiving renal dialysis were affected with Fabry disease [26].
Progressive left ventricular hypertrophy is a common cardiac manifestation of Fabry disease, and the heart may be the only organ affected in the 'cardiac variant' of the disease. It has been estimated that 3% of males suffering from left ventricular hypertrophy and 6% of males with late-onset hypertrophic cardiomyopathy have Fabry disease [27, 28]. Up to 12% of females with late-onset hypertrophic cardiomyopathy may have Fabry disease [29]. Cardiac involvement is extensive and progressive in Fabry disease, and mitral valve prolapse is believed to occur in 50% of affected males [30]. Rolfs and colleagues have also reported a high prevalence of Fabry disease in acute stroke patients: from a cohort of 721 patients, biologically significant mutations were identified in 4.9% of males and 2.4% of females [31].
The majority of reports of Fabry disease are in Caucasians, but isolated cases have been reported in Asian populations. The results of a survey in Japan estimated the frequency of Fabry disease to be 1 in 200 000 [32]. In this study, renal failure was detected in 43% of Fabry patients over the age of 30 years, while cardiac disease was present in 60% of patients over the age of 40 years. Two point mutations have been described in Chinese patients with Fabry disease [33, 34].
It is generally believed that the majority of males with Fabry disease experience multiple clinical manifestations and suffer pain throughout their lives. However, although pain is considered the major clinical symptom of Fabry disease, it has been estimated that between 10% and 20% of patients lack this manifestation [35].
Newborn screening for Fabry disease and other LSDs
Newborn screening can be described as a population-based public health programme applied regionally to reduce the morbidity, severity or mortality of specific genetic disorders. Whether we should screen neonates for Fabry disease raises the question of the potential good versus the potential harm that may result from such a programme. The positive and negative outcomes from population-based screening have been debated extensively [36, 37] and many of the same considerations apply to screening for LSDs. For many LSDs, it is clear that early diagnosis and intervention before the onset of irreversible pathology will provide a substantial benefit to the newborn. There are also other potential benefits. Early diagnosis will enable parents to receive genetic counselling and provide them with reproductive choices. In families affected by an LSD, it is not uncommon to have two or more affected children before the first is diagnosed. Early diagnosis, as provided by a newborn screening programme, would also avoid the prolonged and stressful process of diagnosis that is currently experienced by many patients and families. These benefits are primarily directed to the family rather than the newborn, but are nonetheless significant. On the other hand, the potential harm to the parent/newborn relationship that can result from the knowledge that the baby has an incurable disorder and the concept of depriving families of a 'normal' child for the period of time until the child presents clinically must be considered. These are difficult issues to define and quantify but must be addressed by the community before screening can commence.
Newborn screening involves diagnosis of a genetic disease to enable therapy to be given to prevent or slow the expression of the disease. Currently, there are no newborn screening programmes for Fabry disease or any other LSDs; however, a number of technologies are under development [38, 39]. Chamoles et al. [40] has described diagnostic assays using dried blood spots for the detection of α-galactosidase activity against a 4-methylumbelliferyl substrate in the presence of a high concentration of N-acetylgalactosamine (see also Chapter 17). Fuller et al. [41] reported the immunoquantification of α-galactosidase from dry blood spots, which was also diagnostic for Fabry hemizygotes. The capability of this assay to identify heterozygotes was improved when combined with the immunoquantification of saposin C. Heterozygotes and hemizygotes with Fabry disease, and unaffected controls, were able to be separated using urinary lipid profiling that included the glycolipid substrate ceramidetrihexoside (globotriaosylceramide) [42]. Importantly, measurement of ceramidetrihexoside in dry blood spots allowed clear separation of newborn Fabry hemizygotes from unaffected controls [39].
The cost of screening each LSD individually would in most cases be prohibitive, because of their low prevalence. However, several strategies have been proposed to enable the simultaneous screening of multiple LSDs. An estimate of the combined prevalence of LSDs in the Australian population is conservatively given as 1 in 5000, making simultaneous screening for multiple LSDs economically justifiable.
Technology for LSD screening in the newborn population has been the focus of considerable research over the past few years. Lysosomal enzyme activities and protein are measurable in rehydrated dried blood spots. Fluorometric, radiometric, immunochemical (protein profiling) and electrospray ionization tandem mass spectrometry (ESI-MS/MS) assays have been developed. Protein profiling and ESI-MS/MS offer the capability of assaying the products of several enzymes simultaneously (multiplexing). There have been reports of using these two assay types to make direct measurements of both the amount of lysosomal protein and reaction velocities in rehydrated dried blood spots; the assays are readily adaptable to the process of newborn screening [38, 43, 44].
It is now technically possible to detect Fabry hemizygotes in the newborn period, but guidelines need to be developed to address the following issues before implementing general population screening programmes.
- What is the policy regarding the treatment of pre-symptomatic but affected patients?
- Should enzyme replacement therapy be initiated at the time of diagnosis?
- If therapy is initiated in asymptomatic patients, how should its effectiveness be monitored?
The X-linked inheritance and the potential detection of asymptomatic heterozygotes, who may or may not develop a clinical phenotype, will add a special significance to the debate.
Conclusions
Epidemiological data recording the prevalence and incidence of Fabry disease and other LSDs are limited as a result of differences between geographic regions and ethnic populations, and are likely to be underestimates due to missed diagnoses of these rare disorders. Diagnostic methods under development are addressing the problem of detection in high-risk populations, while the introduction of newborn screening for hemizygotes with Fabry disease and other LSDs will provide early and definitive diagnoses for these disorders. This will enable early intervention that, in many cases, is likely to provide substantial benefit to the newborn, as well as an opportunity for the parents to receive genetic counselling. Eventually, universal screening of the newborn population will provide accurate epidemiological data for all LSDs. Accurate data will be essential to understand the natural history of these diseases and the burden they place on individuals, families, societies and healthcare systems. These are important factors that will drive the development and introduction of new therapies for this group of rare disorders.
References
- 1.
- Meikle PJ, Fietz MJ, Hopwood JJ. Diagnosis of lysosomal storage disorders: current techniques and future directions. Expert Rev Mol Diagn. 2004;4:677–91. [PubMed: 15347261]
- 2.
- Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–8. [PubMed: 8703060]
- 3.
- Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P. et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–7. [PubMed: 7637805]
- 4.
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK. et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–5. [PubMed: 9295267]
- 5.
- Ezaki J, Kominami E. The intracellular location and function of proteins of neuronal ceroid lipofuscinoses. Brain Pathol. 2004;14:77–85. [PMC free article: PMC8095780] [PubMed: 14997940]
- 6.
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406:906–10. [PubMed: 10972294]
- 7.
- Hopwood J, Brooks D. An introduction to the basic science and biology of the lysosome and storage diseases. In: Applegarth D, Dimmick J, Hall J, editors. Organelle diseases. Clinical features, diagnosis, pathogenesis and management. London: Chapman and Hall Medical; 1997. p. 7–36.
- 8.
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–54. [PubMed: 9918480]
- 9.
- Rider J, Rider D. Batten disease: past, present, and future. Am J Med Genet. 1988;(Suppl 5):21–6. [PubMed: 3146319]
- 10.
- Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S. et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–6. [PubMed: 10480370]
- 11.
- Dionisi-Vici C, Rizzo C, Burlina AB, Caruso U, Sabetta G, Uziel G. et al. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140:321–7. [PubMed: 11953730]
- 12.
- Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H. et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12:87–92. [PubMed: 14685153]
- 13.
- Ozkara HA, Topcu M. Sphingolipidoses in Turkey. Brain Dev. 2004;26:363–6. [PubMed: 15275696]
- 14.
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C. et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011–17. [PubMed: 16435194]
- 15.
- Arvio M, Autio S, Louhiala P. Early clinical symptoms and incidence of aspartylglucosaminuria in Finland. Acta Paediatr. 1993;82:587–9. [PubMed: 8338996]
- 16.
- Petersen GM, Rotter JI, Cantor RM, Field LL, Greenwald S, Lim JS. et al. The Tay-Sachs disease gene in North American Jewish populations: geographic variations and origin. Am J Hum Genet. 1983;35:1258–69. [PMC free article: PMC1685967] [PubMed: 6650504]
- 17.
- MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–75. [PMC free article: PMC1734754] [PubMed: 11732485]
- 18.
- Beutler E, Grabowski G. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease, 8th edn. New York: McGraw-Hill; 2001. p. 3635–68.
- 19.
- Vallance H, Ford J. Carrier testing for autosomal-recessive disorders. Crit Rev Clin Lab Sci. 2003;40:473–97. [PubMed: 14582604]
- 20.
- Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–3. [PubMed: 3291628]
- 21.
- Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN. et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998;79:69–72. [PubMed: 9738873]
- 22.
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH. et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–16. [PubMed: 10482961]
- 23.
- Linthorst GE, Hollak CE, Korevaar JC, Van Manen JG, Aerts JM, Boeschoten EW. α-Galactosidase A deficiency in Dutch patients on dialysis: a critical appraisal of screening for Fabry disease. Nephrol Dial Transplant. 2003;18:1581–4. [PubMed: 12897098]
- 24.
- Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G. et al. Natural history of Fabry renal disease: influence of α-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81:122–38. [PubMed: 11889412]
- 25.
- Bekri S, Enica A, Ghafari T, Plaza G, Champenois I, Choukroun G. et al. Fabry disease in patients with end-stage renal failure: the potential benefits of screening. Nephron Clin Pract. 2005;101:c33–8. [PubMed: 15886492]
- 26.
- Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A. et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int. 2003;64:801–7. [PubMed: 12911529]
- 27.
- Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ. et al. Prevalence of Anderson–Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–11. [PubMed: 11914245]
- 28.
- Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M. et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–93. [PubMed: 7596372]
- 29.
- Chimenti C, Pieroni M, Morgante E, Antuzzi D, Russo A, Russo MA. et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation. 2004;110:1047–53. [PubMed: 15313943]
- 30.
- Brady R, Grabowski G, Thadhani R. Fabry disease: review and new perspectives. Synermed Communications. 2001:1–8.
- 31.
- Rolfs A, Bottcher T, Zschiesche M, Morris P, Winchester B, Bauer P. et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366:1794–6. [PubMed: 16298216]
- 32.
- Owada M, Kitigawa T. [Lysosomal storage diseases] Nippon Rinsho. 2001;8:317–27. [PubMed: 11808243]
- 33.
- Tse KC, Chan KW, Tin VP, Yip PS, Tang S, Li FK. et al. Clinical features and genetic analysis of a Chinese kindred with Fabry's disease. Nephrol Dial Transplant. 2003;18:182–6. [PubMed: 12480979]
- 34.
- Chen CH, Shyu PW, Wu SJ, Sheu SS, Desnick RJ, Hsiao KJ. Identification of a novel point mutation (S65T) in α-galactosidase A gene in Chinese patients with Fabry disease. Mutations in brief no. 169. Online. Hum Mutat. 1998;11:328–30. [PubMed: 9554750]
- 35.
- Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I. et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767–72. [PubMed: 14505049]
- 36.
- Wilcken B. Ethical issues in newborn screening and the impact of new technologies. Eur J Pediatr. 2003;162(Suppl 1):S62–6. [PubMed: 14618395]
- 37.
- Meikle PJ, Dean CJ, Brooks DA, Hopwood JJ. Newborn screening for lysosomal storage disorders: ethical and technical considerations. Italian J Pediatr. 2004;30:305–11.
- 38.
- Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F. et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50:1785–96. [PMC free article: PMC3428798] [PubMed: 15292070]
- 39.
- Meikle PJ, Ranieri E, Simonsen H, Rozaklis T, Ramsay SL, Whitfield PD. et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114:909–16. [PubMed: 15466084]
- 40.
- Chamoles NA, Blanco MB, Gaggioli D, Casentini C. Hurler-like phenotype: enzymatic diagnosis in dried blood spots on filter paper. Clin Chem. 2001;47:2098–102. [PubMed: 11719472]
- 41.
- Fuller M, Lovejoy M, Brooks DA, Harkin ML, Hopwood JJ, Meikle PJ. Immunoquantification of α-galactosidase: evaluation for the diagnosis of Fabry disease. Clin Chem. 2004;50:1979–85. [PubMed: 15364892]
- 42.
- Fuller M, Sharp PC, Rozaklis T, Whitfield PD, Blacklock D, Hopwood JJ. et al. Urinary lipid profiling for the identification of Fabry hemizygotes and heterozygotes. Clin Chem. 2005;51:688–94. [PubMed: 15695328]
- 43.
- Umapathysivam K, Hopwood JJ, Meikle PJ. Determination of acid α-glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin Chem. 2001;47:1378–83. [PubMed: 11468225]
- 44.
- Wang D, Eadala B, Sadilek M, Chamoles NA, Turecek F, Scott CR. et al. Tandem mass spectrometric analysis of dried blood spots for screening of mucopolysaccharidosis I in newborns. Clin Chem. 2005;51:898–900. [PubMed: 15695324]
Tables
Table 1Incidence/prevalence of lysosomal storage diseases
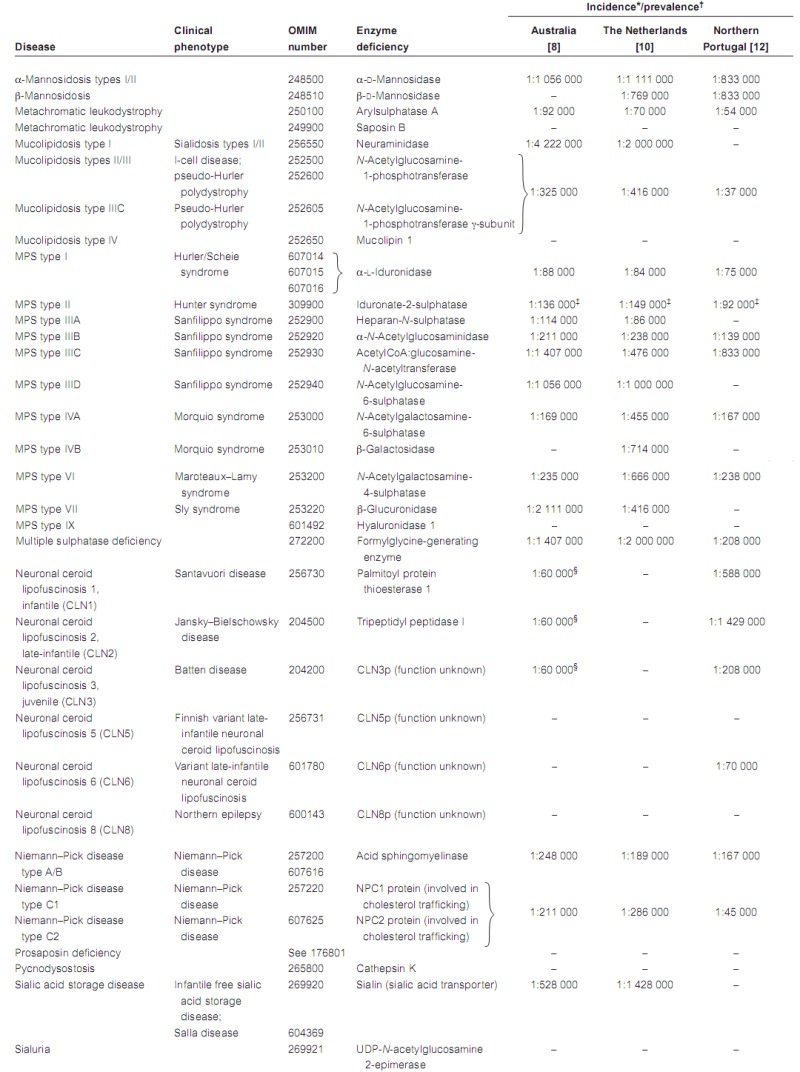
LAMP-2, lysosome-associated membrane protein 2; MPS, mucopolysaccharidosis; OMIM, Online Mendelian Inheritance in Man.
*Incidence in Australia defined as the total number of cases diagnosed within a certain period of time divided by the total number of births in the same period.
- †
Birth prevalence in The Netherlands and Portugal defined as the total number of diagnosed cases born within a certain period of time divided by the total number of births in the same period.
- ‡
Values for hemizygotes only.
- §
Combined incidence of CLN1, CLN2 and CLN3 [1].