The availability of enzyme replacement therapy (ERT) and the possibility of improved organ function, quality of life and ultimately life expectancy has stimulated reevaluation of the clinical expression of Fabry disease in females. Recent editions of general medical textbooks now recognize the burden of signs and symptoms found in heterozygotes. The use of the term carrier in this disorder has been questioned. FOS – the Fabry Outcome Survey – has reinforced the high prevalence of disease manifestations occurring in heterozygous females and now provides a mechanism for evaluating the natural history of disease and effects of ERT with agalsidase alfa in a large cohort of female patients.
Introduction
The realization that females heterozygous for mutations in the α-galactosidase A gene may experience significant manifestations of Fabry disease is relatively recent. Prior to the development of enzyme replacement therapy (ERT), medical literature relating to females with Fabry disease was confined to case reports, and small cohort studies and these were limited by preconceptions implicit in the notion of carrier status in an X-linked recessive condition. Disease-specific symptoms were said to be the exception [1, 2], often with a frequency of significant end-organ damage of no more than 1% in the female heterozygous population [3].
Until recently, general medical textbooks have emphasized that females are largely asymptomatic, citing the seminal article by Desnick et al. [3]. For example, it has been stated that: "Some heterozygous females… experience mild manifestations such as painful neuropathy, as well as other features of the disease" [4]. The following statements appear in another well-known general textbook: "heterozygous female subjects are usually asymptomatic or exhibit mild manifestations"; "corneal opacities and characteristic lenticular lesions are present in about 70 to 80 percent of asymptomatic heterozygotes"; "heterozygous female subjects may have corneal opacities, isolated skin lesions"; and "rare female heterozygotes may have manifestations as severe as those in affected male subjects" [5]. More recently published textbooks, however, reflect the changing view of the expression of Fabry disease in females. The most recent edition of the Oxford Textbook of Medicine Reports: "An unusual feature of Fabry's disease is the presence of clinical signs and symptoms in the majority of heterozygous female carriers of the condition, although these manifestations are usually less severe and of later onset than in affected hemizygous males" [6].
This changing view is reflected in an on-line updated version of Harrison's Principles of Internal Medicine, where in May 2005 it is written that: "Up to 70% of heterozygous females may exhibit clinical manifestations, including central nervous system and cardiac disease, but usually do not develop renal failure".
The peer-reviewed literature, recently stimulated by new prospects of therapy, has systematically documented disease expression in females. This chapter briefly reviews the current literature and describes the part played by FOS – the Fabry Outcome Survey – in defining the effects of Fabry disease in a large cohort of female patients, and its potential for assessing the effects of ERT.
Evidence for the effects of Fabry disease in female patients
Questionnaire studies of extensive cohorts of patients with Fabry disease have provided insight into the range of symptoms that female heterozygotes may experience [7, 8]. More recently, the introduction of ERT has demanded a thorough baseline assessment of all patients, including heterozygous females [9, 10]. It has now become clear that females may exhibit a range of severe disease manifestations, similar to that seen in male hemizygotes (Table 1). Heterozygous females should therefore be regarded as potential patients and not simply as carriers. Whybra and co-workers have gone so far as to postulate that this lysosomal storage disease should be considered as an X-linked dominant disease [9].
Table 1
Literature review of the manifestations of Fabry disease in heterozygous female patients.
In a study of 60 obligate female carriers in a UK registry, MacDermot et al. found that 30% exhibited multiple and serious disease manifestations, including transient ischaemic attacks, stroke and renal failure [7]. No patient investigated by Mehta et al. [10] or Whybra et al. [9] was entirely without clinical manifestations, and some symptoms were found in over 90% of patients.
Neuropathic pain
Neuropathic pain is reported to be a severe, disabling and common feature of Fabry disease in females. Most describe it as continuous, with exacerbations during illness and hot weather. Along with fatigue, neuropathic pain has a significant impact on quality of life (QoL). It has been reported in female patients as young as 4 years of age [9], with a median age of onset of 10 years, and may continue throughout life.
Quality of life
An evaluation conducted in Mainz showed that organ involvement in female patients with Fabry disease has a significant impact on QoL. Before initiating ERT, 15 female patients were asked to complete the short form 36 (SF-36) QoL questionnaire. The results were compared with those from a general German population and from patients with different chronic diseases. Female patients with Fabry disease had SF-36 scores that were substantially lower than those for the general female population. Female patients with Fabry disease also scored substantially lower than females with rheumatoid arthritis in 'general health', 'vitality', 'role emotional' and 'mental health' domains [11].
Cerebrovascular complications
Cerebrovascular complications of stroke and transient ischaemic attacks have been documented in 5–27% of heterozygous females [7, 8, 10], with one study finding them more frequently in female than in male patients (27% compared with 12%) [10]. Mitsias and Levine reported ten heterozygotes with cerebrovascular complications, such as memory loss, dizziness, ataxia, hemiparesis, loss of consciousness and hemisensory disturbance [12]. Corresponding changes on magnetic resonance imaging (MRI), including diffuse white matter disease and infarction of the brainstem and thalamus, have been described [13–15]; however, Morgan et al. failed to find any changes on brain MRI in younger females [16]. Other neurovascular manifestations, including vertigo, tinnitus, hearing loss and hypohidrosis, are reported in approximately one-third of female patients, and Galanos et al. suggested that the presence of anhidrosis in females is predictive of later significant renal disease [8].
Renal involvement
Renal failure is a significant cause of premature death in male hemizygous patients with Fabry disease. In contrast, whilst proteinuria [17] and a reduction in renal function are well described in heterozygotes [18–20], progression to end-stage renal failure is infrequent, with only 1–2% of females requiring dialysis or transplantation [7, 10]. Histological evidence of renal changes has been found not only in those females with evidence of renal dysfunction but also in asymptomatic heterozygotes undergoing investigations as potential kidney donors [21]. Kriegsmann and co-workers published a case report of a 26-year-old female patient who was admitted to hospital because of fever of unknown origin and renal failure [22]. Extracapillary proliferative (crescentic) glomerulonephritis and granulomatous interstitial nephritis were identified by histological, immunohistochemical and electron microscopic analysis of a kidney biopsy, and Fabry disease was confirmed by further investigations [22].
Cardiac symptoms
Cardiac symptoms and evidence of structural cardiac disease, including septal hypertrophy, cardiomyopathy and mitral valve insufficiency, were reported by MacDermot et al. [7] and Whybra et al. [9]. Others have described palpitations in approximately one-third of females [8]. Arrhythmias and a requirement for pacemaker insertion have also been described [23], and Cantor and co-workers reported a 53-year-old female with restrictive cardiomyopathy requiring cardiac transplantation [24].
Gastrointestinal symptoms
Symptoms of diarrhoea, abdominal pain, constipation and vomiting, similar to those seen in irritable bowel syndrome, are described in 50–60% of females (Table 1). The aetiology of gastrointestinal disease is not known. However, the finding of electron-dense sphingolipids in neuronal and vascular tissue of the small bowel [25] suggests that it may be neurovascular in origin.
Mortality
The impact of Fabry disease on female survival remains unclear due to bias in ascertainment. Kaplan–Meier analysis of UK data provided by relatives and confirmed by examination of death certificates suggests a median cumulative survival of 70 years, some 15 years shorter than the reference UK population [7]. A study of deaths in 24 female relatives of patients with Fabry disease in FOS suggested a mean age of death of 55.4 ± 14.9 years, with cardiac disease being the most frequent cause [10].
FOS data
Patient demographics and clinical features
Data are currently available from 358 female patients recruited to the FOS database. The mean age at entry into FOS was 37.5 ± 18.3 years. Diagnosis was made at a mean age of 31.4 ± 17.1 years (n = 316), while the mean age at onset of symptoms was 19.1 ± 15.0 years (n = 214). Eighteen females did not have any symptoms reported (mean age at latest clinic visit, 26.1 ± 18.0 years). Patients have been recruited from 52 centres in 11 European countries.
The frequency and age at onset of disease-specific clinical features, as reported by the patient using a checklist of predefined clinical features developed for FOS, are shown in Table 2. The most frequent features are neurological and cardiac, which are reported in 67% and 51% of patients, respectively. Neurological features are also the earliest to develop, beginning at an average age of 21.9 years, whereas cardiac features begin later, at an average age of 36.6 years. Self-reported indicators of renal involvement are recorded in 38% of patients and begin at an average age of 37.4 years. Other signs and symptoms involving the eye and gastrointestinal system are also common, being observed in nearly 50% of the females for whom these data are available.
Table 2
Frequency and age at onset of specific signs and symptoms of Fabry disease in heterozygous female patients enrolled in FOS – the Fabry Outcome Survey.
When the signs and symptoms are analysed in more detail, the predominant symptoms reported are acute attacks of pain (classed as neurological), affecting 52% of patients. The next most frequent features are angiokeratoma and cornea verticillata. A high proportion of patients (34%) are also reported to have proteinuria. Of the cardiac manifestations reported, left ventricular hypertrophy is found in 26% of the female population studied, and palpitations are reported by 21%.
Clinical investigations
Echocardiographic assessment of left ventricular mass (LVM) prior to starting ERT was available in 151 female patients in FOS. The mean LVM index (± SD) was 50.4 ± 25.7 g/m2.7 at a mean age of 39.5 ± 17.6 years. Hypertrophic cardiomyopathy was considered to be present if the LVM index exceeded 50 g/m2.7. Mean ventricular wall thickness was 10.9 ± 3.3 mm. The mean glomerular filtration rate (GFR) in 245 female patients enrolled in FOS prior to ERT was 77.1 ± 17.5 ml/min/1.73 m2 at a mean age of 43.1 ± 14.6 years, excluding those below 18 years of age. Classification of renal function according to the Kidney Disease Outcomes Quality Initiative guidelines shows that, among females in FOS, 13% have stage III (moderate), 64% have stage II (mild) and 22% have stage I (normal) renal function [26].
Proteinuria
A correlation between age and GFR was not seen in a subgroup of 41 female patients with proteinuria (protein > 300 mg/24 hours). The mean GFR was 70.1 ± 17.7 ml/min/1.73 m2 at ages ranging from 17 to 71 years.
Disease severity and progression in untreated female patients
The LVM index (Figure 1) increases exponentially with age (r = 0.78, p < 0.001, n = 151), and the mean ventricular wall thickness increases in a more linear fashion with age (r = 0.78, p < 0.001, n = 164). Age is negatively correlated with estimated GFR (assessed using the Modification of Diet in Renal Disease [MDRD] equation), with a decline in GFR of 0.62 ml/min/year (Figure 2; r = −0.54, p < 0.001, n = 245). In addition, age is negatively correlated with health-related QoL; however, there is no correlation after an adjustment has been made for the expected normal decline in QoL with age. 'Pain at its least' (r = 0.26, p = 0.001, n = 195) and 'pain right now' (r = 0.23, p < 0.001, n = 195), assessed using the Brief Pain Inventory, correlated positively with age. No significant correlation with age was found in relation to 'pain at its worst' (r = 0.10, p = 0.15, n = 195) or 'pain on average' (r = 0.12, p = 0.10, n = 195).
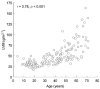
Figure 1
Correlation between left ventricular mass (LVM) index and age in females with Fabry disease (n = 151) enrolled in FOS – the Fabry Outcome Survey.
![Figure 2. Correlation between estimated glomerular filtration rate (eGFR, assessed using the Modification of Diet in Renal Disease [MDRD] equation) and age in females with Fabry disease (n = 245) enrolled in FOS – the Fabry Outcome Survey.](/books/NBK11591/bin/ch30f2.gif)
Figure 2
Correlation between estimated glomerular filtration rate (eGFR, assessed using the Modification of Diet in Renal Disease [MDRD] equation) and age in females with Fabry disease (n = 245) enrolled in FOS – the Fabry Outcome Survey.
The modification of the Mainz Severity Score Index for FOS (FOS-MSSI) correlates positively with age (Figure 3; r = 0.46, p < 0.001, n = 337). It correlates inversely, however, with health-related QoL, as assessed using the European QoL (EQ-5D) utility score (r = −0.41, p < 0.001, n = 134), a measure of the deviance of the EQ-5D scores from age- and gender-matched UK reference data [27–29] (r = −0.34, p < 0.001, n = 134), and with the visual analogue scale of the EQ-5D (r = −0.42, p < 0.001, n = 111).
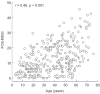
Figure 3
Correlation between the modification of the Mainz Severity Score Index for the Fabry Outcome Survey (FOS-MSSI) and age in females with Fabry disease (n = 337) enrolled in FOS.
Mortality
Mortality data, according to patient recall, are available for some relatives believed to have Fabry disease. The mean age at death in 39 female relatives was 57.8 ± 14.3 years (Figure 4).
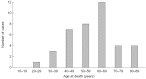
Figure 4
Mortality data for female relatives of patients in FOS – the Fabry Outcome Survey – believed to have had Fabry disease (according to patient recall).
Ascertainment bias
A potential limitation of the FOS database relates to ascertainment bias. The numbers of females and males within FOS are currently approximately equal. Theoretical considerations suggest that, assuming equal relative fecundity of males and females, heterozygotes should outnumber hemizygotes by two to one. We hypothesize that the shortfall is due to the relative under-representation of unaffected females in FOS. A secular trend has become apparent over the first 5 years of FOS. Initially, when males outnumbered females, the severity of disease in females enrolled in FOS was higher than is now the case, as more young and asymptomatic women are recruited (see Chapter 16).
Relationship between X-inactivation, enzyme levels and disease severity
Current understanding of disease expression in females with X-linked disorders suggests that the severity of manifestations depends on the degree to which the normal X-chromosome is inactivated. Random skewing of X-inactivation may result in variation of expression of 25–75% of normal enzyme levels and, in cases of more severe or non-random skewing, expression levels of less than 25% may be seen. Preliminary studies by Morrone et al. have shown that there may be a correlation between skewing of X-inactivation in peripheral blood leukocytes and clinical expression of Fabry disease [30]. This finding requires confirmation. If this were the only factor, one might predict a straightforward relationship between blood enzyme activity and disease severity. Data on plasma or leukocyte enzyme activity are available from 243 patients in FOS. Cross-tabulation with disease severity, as determined by the FOS-MSSI, demonstrated that among 96 patients with normal enzyme activity, 39% had a score above 20 (moderate disease severity), whereas 26 out of 147 patients (18%) with reduced enzyme activity had a score above 20. Thus, data from FOS do not support the use of enzyme activity in females as a marker of disease severity.
There are two X-linked disorders involving deficiency of lysosomal enzymes: Fabry disease and Hunter syndrome. In Hunter syndrome, clinical disease expression is exceptionally rare in females and, where observed, has been related to extreme Lyonization in almost all cases. This would suggest that the low levels of enzyme that are synthesized and secreted in all but extreme cases are sufficient to cross-correct deficient synthesis by abnormal cells. Cross-correction has been demonstrated in Hunter cells in vitro. This has not as yet been demonstrated in Fabry disease, and the fact that females with normal plasma enzyme activity may manifest moderately severe disease suggests that uptake of normal enzyme by abnormal cells may be defective. The clinical improvement achieved by ERT described below suggests, on the other hand, that cellular uptake of α-galactosidase A is possible in vivo in female patients.
ERT in females
The general effects of ERT are described later in this volume. ERT was initially evaluated in clinical trials of male patients. Bähner and co-workers evaluated the safety, efficacy and pharmacokinetics of agalsidase alfa (Replagal®; TKT Europe AB) administered intravenously to female patients with Fabry disease in an open-label, single-centre study [11]. Agalsidase alfa was shown to be well tolerated, and none of the female patients developed antibodies or experienced infusion reactions. The pharmacokinetic profile of agalsidase alfa in female patients is comparable to the pharmacokinetics in male patients. Mean levels of globotriaosylceramide – the lysosomal storage product in Fabry disease – in urinary sediment and plasma had decreased from baseline after 13, 27 and 41 weeks of ERT. A significant decrease in LVM from baseline was seen at weeks 27 (p = 0.003) and 41 (p = 0.039), and a significant reduction in the QRS duration was seen at week 27 (p = 0.007). Furthermore, there was a significant improvement in QoL. Renal function did not deteriorate in these 15 female patients over the 13–41 weeks of observation. It was concluded that ERT with agalsidase alfa was safe and effective in heterozygous females with Fabry disease [11].
Phenotypic heterogeneity and the lack of biomarkers have hindered the monitoring of responses to ERT. Whybra and co-workers therefore developed a scoring system – the MSSI – to measure the severity of Fabry disease and to monitor the clinical course of the disease in response to ERT [31]. Most male patients with Fabry disease were rated as severely or moderately affected on the MSSI, whereas most female patients were rated as moderately affected [31]. Although there was a trend towards a higher (worse) general score in males when compared with females (p = 0.078), this was no longer apparent after ERT. After 1 year of ERT, QoL had improved more in males than in females [32]. In those women with impaired health-related QoL (EQ-5D < 1), ERT was associated with an improvement in this important measure of outcome after 1 year of treatment (signed rank-sum test, p < 0.05, n = 38). This was equally the case when adjusted for age-related trends in QoL.
Analysis of organ-specific data from patients in FOS has demonstrated that cardiac structure and function improves and renal function is stabilized by ERT with agalsidase alfa (see Chapters 37 and 38). Analysis of the long-term effects of ERT in women is currently limited by the availability of follow-up data.
Conclusions
FOS has allowed investigators in many countries to pool their experience of patients with Fabry disease. In the case of disease expression in women, FOS has provided data to refute the long-held assumption that women with Fabry disease are rarely symptomatic. The data within FOS will be of immense value in addressing important issues relating to the pathogenesis of Fabry disease in women and their response to ERT.
References
- 1.
- Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M. et al. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–93. [PubMed: 7596372]
- 2.
- Redonnet-Vernhet I, Ploos van Amstel JK, Jansen RP, Wevers RA, Salvayre R, Levade T. Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the α-galactosidase A gene. J Med Genet. 1996;33:682–8. [PMC free article: PMC1050704] [PubMed: 8863162]
- 3.
- Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. 8th edn. New York: McGraw-Hill; 2001. p. 3733–74.
- 4.
- Jameson L, Kopp P. Principles of human genetics. In: Kasper DL, Braunwald E, Fauci A, Hauser S, Longo D, Jameson JL, editors. Harrison's priciples of internal medicine. 16th edn. New York: McGraw-Hill; 2004.
- 5.
- McGovern M, Desnick R. Chapter 217. In: Goldman L, Ausiello D, editors. Cecil textbook of medicine. 22nd edn. Philadelphia: Elsevier; 2003. p. 1278–9.
- 6.
- Cox TM. Metabolic disorders: lysosomal storage diseases. In: Warrell DA, Cox TM, Firth JD, Benz EJ, editors. Oxford textbook of medicine. 4th edn. Oxford: Oxford University Press; 2003. p. 119–120 and 416.
- 7.
- MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–75. [PMC free article: PMC1734754] [PubMed: 11732485]
- 8.
- Galanos J, Nicholls K, Grigg L, Kiers L, Crawford A, Becker G. Clinical features of Fabry's disease in Australian patients. Intern Med J. 2002;32:575–84. [PubMed: 12512750]
- 9.
- Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J. et al. Anderson–Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24:715–24. [PubMed: 11804208]
- 10.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C. et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–42. [PubMed: 15025684]
- 11.
- Bähner F, Kampmann C, Whybra C, Miebach E, Wiethoff CM, Beck M. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis. 2003;26:617–27. [PubMed: 14707510]
- 12.
- Mitsias P, Levine SR. Cerebrovascular complications of Fabry's disease. Ann Neurol. 1996;40:8–17. [PubMed: 8687196]
- 13.
- Grewal RP, McLatchey SK. Cerebrovascular manifestations in a female carrier of Fabry's disease. Acta Neurol Belg. 1992;92:36–40. [PubMed: 1546524]
- 14.
- Castro LH, Monteiro ML, Barbosa ER, Scaff M, Canelas HM. Fabry's disease in a female carrier with bilateral thalamic infarcts: a case report and a family study. Rev Paul Med. 1994;112:649–53. [PubMed: 7481431]
- 15.
- Hasholt L, Sorensen SA, Wandall A, Andersen EB, Arlien-Soborg P. A Fabry's disease heterozygote with a new mutation: biochemical, ultrastructural, and clinical investigations. J Med Genet. 1990;27:303–6. [PMC free article: PMC1017080] [PubMed: 2161929]
- 16.
- Morgan SH, Rudge P, Smith SJ, Bronstein AM, Kendall BE, Holly E. et al. The neurological complications of Anderson–Fabry disease (α-galactosidase A deficiency) – investigation of symptomatic and presymptomatic patients. Q J Med. 1990;75:491–507. [PubMed: 2167495]
- 17.
- Yuen NW, Lam CW, Chow TC, Chiu MC. A characteristic dissection microscopy appearance of a renal biopsy of a Fabry heterozygote. Nephron. 1997;77:354–6. [PubMed: 9375832]
- 18.
- Rodriguez FH Jr,, Hoffmann EO, Ordinario AT Jr,, Baliga M. Fabry's disease in a heterozygous woman. Arch Pathol Lab Med. 1985;109:89–91. [PubMed: 2982342]
- 19.
- Van Loo A, Vanholder R, Madsen K, Praet M, Kint J, De Paepe A. et al. Novel frameshift mutation in a heterozygous woman with Fabry disease and end-stage renal failure. Am J Nephrol. 1996;16:352–7. [PubMed: 8739292]
- 20.
- el-Shahawy MA, Mesa C, Koss M, Campese VM. A 19-year-old female with fever, acroparesthesia, and progressive deterioration of renal function. Am J Nephrol. 1996;16:417–24. [PubMed: 8886180]
- 21.
- Gubler MC, Lenoir G, Grunfeld JP, Ulmann A, Droz D, Habib R. Early renal changes in hemizygous and heterozygous patients with Fabry's disease. Kidney Int. 1978;13:223–35. [PubMed: 418264]
- 22.
- Kriegsmann J, Otto M, Wandel E, Schwarting A, Faust J, Hansen T. et al. [Fabry's disease, glomerulonephritis with crescentic and granulomatous interstitial nephritis. Case of one family] Pathologe. 2003;24:439–43. [PubMed: 14605848]
- 23.
- Nakayama Y, Tsumura K, Yamashita N, Yoshimaru K. Dynamic left ventricular arterial pressure gradient and sick sinus syndrome with heterozygous Fabry's disease improved following implantation of a dual chamber pacemaker. Pacing Clin Electrophysiol. 1999;22:1114–15. [PubMed: 10456648]
- 24.
- Cantor WJ, Daly P, Iwanochko M, Clarke JT, Cusimano RJ, Butany J. Cardiac transplantation for Fabry's disease. Can J Cardiol. 1998;14:81–4. [PubMed: 9487277]
- 25.
- Sheth KJ, Werlin SL, Freeman ME, Hodach AE. Gastrointestinal structure and function in Fabry's disease. Am J Gastroenterol. 1981;76:246–51. [PubMed: 6274188]
- 26.
- National Kidney Foundation (NKF) Kidney Disease Outcome Quality Initiative (K/DOQI) Advisory Board. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39(Suppl 2):S1–246. [PubMed: 11904577]
- 27.
- Kind P, Hardman G, Macran S. UK population norms for EQ-5D. York Centre for Health Economics discussion paper 1999;172.
- 28.
- van Agt HM, Essink-Bot ML, Krabbe PF, Bonsel GJ. Test-retest reliability of health state valuations collected with the EuroQol questionnaire. Soc Sci Med. 1994;39:1537–44. [PubMed: 7817218]
- 29.
- Brooks R. Quality of life measures. Crit Care Med. 1996;24:1769. [PubMed: 8874318]
- 30.
- Morrone A, Cavicchi C, Bardelli T, Antuzzi D, Parini R, Di Rocco M. et al. Fabry disease: molecular studies in Italian patients and X inactivation analysis in manifesting carriers. J Med Genet. 2003;40:e103. [PMC free article: PMC1735554] [PubMed: 12920095]
- 31.
- Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E. et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson–Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004;65:299–307. [PubMed: 15025723]
- 32.
- Whybra C, Wendrich K, Ries M, Gal A, Beck M. Clinical manifestation in female Fabry disease patients. Contrib Nephrol. 2001:245–50. [PubMed: 11688388]
Publication Details
Author Information and Affiliations
Authors
Patrick B Deegan,1 Frank Bähner,2 Miguel Barba,3 Derralynn A Hughes,4 and Michael Beck2.Affiliations
Copyright
Publisher
Oxford PharmaGenesis, Oxford
NLM Citation
Deegan PB, Bähner F, Barba M, et al. Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 30.

![Figure 2. Correlation between estimated glomerular filtration rate (eGFR, assessed using the Modification of Diet in Renal Disease [MDRD] equation) and age in females with Fabry disease (n = 245) enrolled in FOS – the Fabry Outcome Survey.](/books/NBK11591/bin/ch30f2.jpg)

