

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Chemical Agents and Related Occupations. Lyon (FR): International Agency for Research on Cancer; 2012. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 100F.)

Two of these agents (2,3,7,8-tetrachlorodibenzo-para-dioxin and 2,3,4,7,8-pentachlorodibenzofuran were specifically considered by previous IARC Working Groups in 1977, 1987, and 1997 (IARC, 1977, 1987, 1997). The Working Group in 1987 reviewed polychlorinated biphenyls, but did not specifically consider 3,3′,4,4′,5-pentachlorobiphenyl. Since the previous evaluations new data have become available, which have been incorporated in this Monograph, and taken into consideration in the present evaluation.
1. Exposure Data
1.1. Identification of the agents
From NTP (2006a, b, c)
1.1.1. 2,3,7,8-Tetrachlorodibenzo-para-dioxin (2,3,7,8-TCDD, TCDD)
- Chem. Abstr. Serv. Reg. No.: 1746-01-6
- Chem. Abstr. Serv. Name: 2,3,7,8-Tetrachlorodibenzo[b,e][1,4]dioxin
- Synonyms: 2,3,7,8-TCDD; TCDD; dioxin; tetradioxin
- C12H4Cl4O2
- Relative molecular mass: 321.98
- Description: Colourless to white crystalline solid
- Solubility: Insoluble in water; slightly soluble in n-octanol and methanol; and soluble in other organic solvents (e.g. dichlorobenzene, chlorobenzene, benzene, chloroform, and acetone)
- Octanol/water partition coefficient: log Kow, 6.80
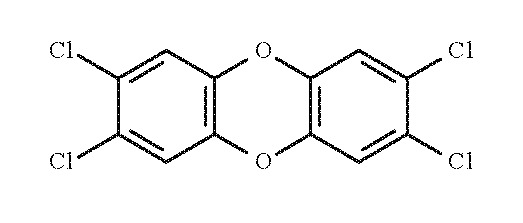
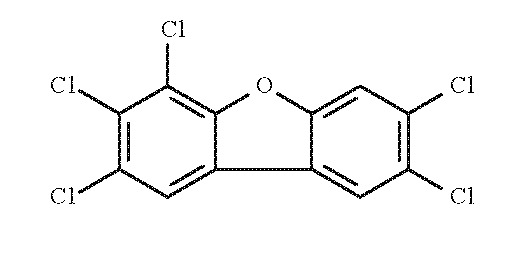
1.1.2. 2,3,4,7,8-pentachlorodibenzofuran (2,3,4,7,8-PeCDF)
- Chem. Abstr. Serv. Reg. No.: 57117-31-4
- Chem. Abstr. Serv. Name: 2,3,4,7,8-Pentachlorodibenzofuran
- Synonym: 2,3,4,7,8-PeCDF; 2,3,4,7,8-penta-CDF
- C12H3Cl5O
- Relative molecular mass: 340.42
- Description: Solid with a melting point of 195–196 °C (NTP Chemical Repository Information). It is stable under normal laboratory conditions.
- Solubility in water: 2.36 × 10−4 mg/L at 22.7 °C
- Octanol/water partition coefficient: log Kow, 6.92
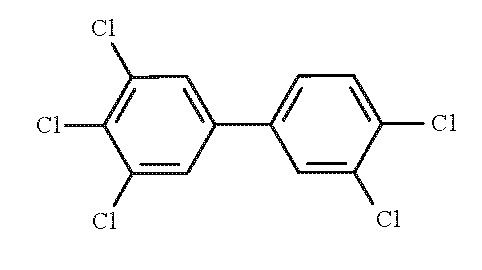
1.1.3. 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126)
- Chem. Abstr. Serv. Reg. No.: 57465-28-8
- Chem. Abstr. Serv. Name: 3,3′,4,4′,5-Pentachlorobiphenyl
- Synonym: PCB 126
- C12H5Cl5
- Relative molecular mass: 326.42
- Description: Solid with a melting point of 160–161 °C.
- Solubility in water: 1.03 × 10−3 mg/L at 25 °C
- Octanol/water partition coefficient: log Kow, 6.89
1.2. Occurrence and use
2,3,7,8-Tetrachlorodibenzo-para-dioxin (TCDD) has no known commercial applications. It occurred as a contaminant in chlorophenoxy herbicides, including 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), which were widely used in the 1960s and 1970s to control weeds (e.g. on pastureland and food crops) and as a defoliant during the Viet Nam war. It is used as a research chemical and was tested, but never used commercially, as a flame-proofing agent and as a pesticide against insects and wood-destroying fungi (NTP, 2004). TCDD may also be produced in thermal processes such as incineration, in metal-processing, and in the bleaching of paper pulp with free chlorine. The relative amounts of the TCDD congeners produced depend on the production or incineration process and vary widely (IARC, 1997).
Polychlorinated dibenzofurans (PCDFs) are not manufactured commercially other than for scientific research purposes. Release of PCDF into the environment is mainly from combustion and incineration. Based on congener-specific profiles, combustion sources all produce 2,3,7,8-substituted polychlorinated dibenzo-para-dioxins (PCDDs) and PCDFs, including 2,3,4,7,8-pentachlorodibenzofuran (PeCDF). The latter substance is the major congener emitted from cement kilns burning hazardous waste (approximately 20% of the total congener emission). Other major sources of PeCDF are metal smelting, refining, and processing; chemical manufacturing/processing (production of chlorophenols, PCBs, vinyl chloride); pulp bleaching; and existing reservoirs that reflect past releases (IARC, 1997; USEPA, 2000a).
Mixtures of polychlorobiphenyls (PCBs), including 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126), were produced for commercial purposes during the period 1929–1977 for the electrical industry, to be used as dielectric insulating fluids for transformers and capacitors. PCBs were also used in hydraulic fluids, plastics, and paints. The manufacture and use of PCBs in the United States was stopped in 1977 in view of increasing amounts of PCB residues in the environment that had accumulated during the previous decades. However, PCBs continue to be released into the environment through the use and disposal of products containing PCBs, as by-products during the manufacture of certain organic chemicals, and during combustion of some waste materials (USEPA, 2000a). Due to their lipophilic nature (log Kow, 6.5–7.7) and resistance to biodegradation, specific PCBs bio-concentrate and bio-accumulate in the environment. PCBs are widespread in their distribution and are found in virtually all environmental compartments including air, soil, water, sediment, and biota (USEPA, 2000b).
1.3. Human exposure
PCDDs are ubiquitous in soil, sediments and air. Excluding occupational or accidental exposures, most human exposure to TCDD occurs as a result of eating meat, milk, eggs, fish and related products, as TCDD is persistent in the environment and accumulates in animal fat. Occupational exposures to TCDD at higher levels have occurred since the 1940s as a result of production and use of chlorophenols and chlorophenoxy herbicides. Even higher exposures have occurred sporadically in relation to accidents in these industries (IARC, 1997).
Mean background levels of TCDD in human tissues are in the range of 2–3 ng/kg (parts per trillion; ppt) fat [because PCDDs are stored in fat tissue, body burdens of PCDDs are often expressed as concentration in lipid, e.g. 100 ppt lipid]. Available data suggest that these levels have decreased three- to fivefold since the late 1970s, when the development of gas chromatography/mass spectrometry methodology permitted these extremely low concentrations of TCDD in tissues and in the environment to be measured accurately for the first time. Similarly, since the mid-1980s, mean tissue concentrations of total TCDD in the general population have decreased by two- to threefold. Human exposures related to occupation or accidents have led to tissue concentrations of TCDD up to several orders of magnitude higher than background levels (IARC, 1997).
1.3.1 Occupational exposure to dioxins
Because TCDD has never been intentionally manufactured for large-scale commercial use, estimates of the numbers of workers exposed in the past are not available. From the US National Occupational Exposure Survey (1981–1983), it was estimated that approximately 14 workers were potentially exposed to TCDD in the USA (NIOSH, 1990). Historical occupational exposures to TCDD have been reviewed (IARC, 1997).
In a series of studies, Collins et al. (2006, 2007, 2008) examined concentrations of TCDD in serum from 375 workers in Michigan who had been exposed in the past (26–62 years before) to trichlorophenol and/or pentachlorophenol. Workers exposed only to trichlorophenol had mean lipid-adjusted TCDD levels of 15.9 ppt, compared with 6.5 ppt in unexposed workers and 3.3 ppt in community controls. Those exposed to pentachlorophenol only had mean TCDD concentrations of 8.0 ppt; workers exposed to both chemicals had mean TCDD levels of 13.9 ppt; and tradesmen with plant-wide responsibilities had mean levels of 20.7 ppt. A follow-up study to evaluate the influence of various factors on TCDD concentrations in serum of 412 workers exposed to penta- and trichlorophenol, showed that age and body fat were important determinants, whereas diet and jobs-outside-the-plant had little impact (Burns et al., 2008).
To estimate job-specific exposures over time at a facility in New Zealand that manufactured 2,4,5-T along with other products between 1962 and 1988, Aylward et al. (2010) integrated available work records, TCDD pharmacokinetic data, and serum-sampling data from 346 workers. Estimated TCDD concentrations in serum were below 300 ppt lipid for all individuals in the cohort over the entire study period, i.e. lower than estimates for other 2,4,5-T worker populations.
McLean et al. (2009) measured dioxin concentrations in serum of 94 randomly selected former sawmill workers (71 exposed to pentachlorophenol, 23 non-exposed) in New Zealand, twenty years after the use of pentachlorophenol (PCP) had ceased. The authors compared age-adjusted dioxin levels in the exposed and non-exposed groups, examined the effect of duration and intensity of the exposure to PCP, and compared congener profiles with those found in the commercial-grade PCP used at the time. Mean levels in exposed and unexposed workers were 1.88 pg/g (range, 0.51–4.13) and 1.48 pg/g, respectively. The congener profiles in serum were consistent with those in PCP solutions, and dioxin levels increased with both employment duration and estimated exposure intensity.
In a study of Russian workers who manufactured phenoxy herbicides and related compounds in the 1960s, workers who handled 2,4,5-T (n = 34) had median blood-lipid TCDD concentrations of 165.7 ng/kg (parts per trillion), with a range from 35 to 1680 ng/kg. Workers who manufactured 2,4-dichlorophenoxyacetic acid (2,4-D; n = 6) had median levels of 68.9 ng/kg. Children of workers who handled 2,4,5,-T (n = 8) and administrative workers (n = 5) had higher median levels than two control groups drawn from the general population (n = 60): the median blood-lipid TCDD concentrations in the children and administrative staff were 39.5 and 31.0 ng/kg, respectively, compared with 12 and 62.4 ng/kg for the two control groups (Ryan & Schecter, 2000).
Several exposure-response analyses have been performed in the industrial cohorts that served as a basis for the previous IARC Monographs evaluation (Becher et al., 1998; Flesch-Janys et al., 1998; Hooiveld et al., 1998; Steenland et al., 1999, 2004; Crump et al., 2003).
1.3.2 Non-occupational exposure to dioxins
PCDDs, including TCDD, have been measured in all environmental compartments including ambient air, surface water, groundwater, soil, and sediment. While the manufacture and use of chlorinated compounds – chlorophenols, chlorinated phenoxy herbicides – were important sources of PCDD-release into the environment in the past, the restricted manufacture of many of these compounds has substantially reduced their contribution to environmental pollution. It is now recognized that incineration/combustion processes are the most important sources of PCDDs to the environment (Zook & Rappe, 1994; ATSDR, 1998).
The identified sources of environmental release of TCDDs have been grouped into four major categories: incineration sources (municipal waste, hospital waste, hazardous waste, sewage sludge), combustion sources (cement kilns, wood-burning, diesel vehicles, coal-fired utilities, crematoria), industrial sources (pulp and paper mills, chemical manufacturing, metal industry), and reservoir sources (biochemical processes, photolytic processes, forest fires, accidental releases) (Kulkarni et al., 2008).
Human exposure to all dioxin-like compounds is usually calculated in terms of toxic equivalence quotients (TEQs).
Because the various polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, and polychlorinated biphenyls have different activity levels, a toxic equivalence quotient (TEQ) is calculated by standardizing the individual congener levels detected in each sample, multiplying them with the appropriate toxic equivalency factor (TEF), and summing these normalized values. TEFs have been established by the World Health Organization and are calculated relative to 2,3,7,8-TCDD (Van den Berg et al., 2006; Charnley & Kimbrough, 2006; see also Section 4).
The US Environmental Protection Agency (USEPA) reported that emissions from quantified sources – waste incineration, pesticide manufacture, chlorine bleaching of pulp and paper – in the USA decreased from about 14 000 g TEQ/year in 1987 to approximately 1500 g TEQ/year in 2000 (a 90% reduction). This decline is expected to continue (Charnley & Kimbrough, 2006; USEPA, 2006).
People are exposed to PCDDs primarily through foods that are contaminated as a result of the accumulation of these substances in the food-chain and in high-fat foods, such as dairy products, eggs, animal fats, and some fish. Additional exposure sources include industrial accidents (Baccarelli et al., 2002) and several miscellaneous exposures (Yoshimura, 2003; Kulkarni et al., 2008). Because dioxins are fat-soluble, lowering the fat content in food can reduce the intake of dioxin. The average adult in the USA has a daily TEQ intake of approximately 1 pg/kg, lower than a decade ago, whereas a nursing infant has an average TEQ intake of 35–53 pg/kg bw/day (Schecter et al., 1994; USEPA, 2004; Schecter et al., 2006).
Data on TCDD concentrations in lipids have been collected over a 30-year period (1970–2000) among the general population in the USA, Canada, Germany, and France. Mean lipid-levels of TCDD steadily decreased nearly 10-fold over this time period, with lipid-adjusted TCDD concentratrions of about 2 ppt in the year 2000. On the basis of pharmacokinetic modelling, mean concentrations of TCDD in the general population are likely to decrease further to 0.5–1 ppt by 2015, even if intake levels do not decrease further (Aylward & Hays, 2002; Hays & Aylward, 2003).
There is evidence from the NHANES serum PCDD/F data (including TCDD) that concentrations of these compounds in the US population are declining. Median serum-lipid concentrations (population-weighted) of PCDDs/Fs were 13.46, 13.98 and 11.39 TEQ/g lipid for 1999–2000 (n = 1271), 2001–2002 (n = 1244), and 2003–2004 (n = 1290), respectively. When the temporal trends are examined by age, median levels appear to have declined in the younger part of the population and remained essentially constant (or have slightly increased) in the older part (LaKind et al., 2009).
Several exposure studies have shown that some US Viet Nam veterans who were exposed to Agent Orange had serum TCDD levels up to 600 ppt in lipid many years after they had left Viet Nam, compared with values of approximately 1–2 ppt of TCDD for the general population (Kahn et al., 1988; Schecter et al., 1990, 1992; Michalek et al., 1995). In Viet Nam, TCDD levels up to 1 000 000 ppt have been found in soil or sediment from Agent Orange-contaminated areas 3–4 decades after spraying. In addition, elevated concentrations have been measured in food and wildlife in Viet Nam (Olie et al., 1989) as well as in Vietnamese people from contaminated areas (Schecter et al., 2001, 2002, 2003; Dwernychuk et al., 2002; Schecter et al., 2006).
Bates et al. (2004) determined persistent organochlorines, including TCDD, in serum of the non-occupationally exposed New Zealand population in 1996–1997. The weighted mean concentration of TCDD in adult New Zealanders was 2.3 ng/kg lipid weight basis (range, < 1–7.0 ng/kg). The age group-specific data showed a trend towards higher concentrations in the older age groups.
Several recent studies have assessed and reviewed the exposure to and intake of dioxins, including TCDD, from dietary sources (Liem et al., 2000; Tsutsumi et al., 2001; Huwe, 2002; Parzefall, 2002; Baars et al., 2004; Charnley & Doull, 2005; Nakatani et al., 2005; Larsen, 2006; Sasamoto et al., 2006; Gies et al., 2007; Todaka et al., 2008).
1.3.3 Human exposure to polychlorinated dibenzofurans, polychlorinated biphenyls and dioxin-like compounds
Due to high lipophilicity and low solubility in aqueous media, polychlorinated dibenzofurans (PCDFs) accumulate in the fat tissue of animals. The highest concentrations of PCDFs are found in fish, meat, eggs, and dairy products (Schecter et al., 1994; USEPA, 2000b). This results in widespread exposure of the general population to PCDFs and related dioxin-like compounds, with an estimated 90% of human exposure due to ingestion of contaminated food, and a small fraction via inhalation and dermal absorption.
Human exposure to all dioxin-like compounds is usually calculated in terms of TEQs (see definition above). Adult daily intake of dioxin-like compounds including PCDDs, PCDFs and dioxin-like PCBs from all sources is estimated to be approximately 70pg TEQ/day, where TEQ reflects the potency-adjusted amount of all dioxin-like compounds. The intake from all sources of PCDDs and PCDFs is estimated to be 45pg TEQ/day and intake from dioxin-like PCBs is 25pg TEQ/day. Approximately 90% of the daily intake is from food sources (40pg TEQ/day for PCDDs and PCDFs and 22pg TEQ/day for dioxin-like PCBs). Intake of PeCDF from food is approximately 6.6 pg per day and represents 16% of the total intake of PCDDs and PCDFs on a TEQ basis. This level of exposure together with the long half-life of dioxin-like compounds in humans leads to persistent body burdens in humans in the range of 25 pg TEQ/g lipid (USEPA, 2000b). Depending upon dietary practice and proximity to specific sources of exposure, some populations may have higher exposure or body burdens. In contrast to the general population, several specific groups may have been exposed to much higher levels of PeCDF as a result of occupational exposure. In European tissue samples, PeCDF levels are on average 13 ppt TEQ (lipid-adjusted) and represent approximately 36% of the TEQ contributed by PCDDs and PCDFs. PeCDF is the highest contributor of the PCDF class of DLCs to the total TEQ, based on both intake levels and tissue concentrations.
The majority (90%) of ambient human exposure to dioxin-like compounds occurs through the ingestion of food containing PCB residues. Levels of PCB 126 in food range from 0.05 to 0.83 pg/g. Human exposure to all dioxin-like compounds is usually calculated in terms of toxic equivalents (TEQs). On a TEQ basis, it is estimated that humans are exposed via food to 22 pg TEQ/day (for a 70-kg person) from dioxin-like PCBs of which PCB 126 (13 pg/day) accounts for 60% of the TEQ intake. Bioaccumulation of PCBs results in persistent levels of these substances in human tissues. With an average concentration of 12 pg TEQ/g lipid, PCB 126 accounts for approximately half of the PCB TEQ in human tissues (USEPA, 2000b).
2. Cancer in Humans
Human exposures related to occupation or accidents have led to tissue concentrations of 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) that are 10–100-fold higher than average background levels in the 1980s. The highest exposures occurred in industrial cohorts of workers producing phenoxy herbicides and chlorophenols, while exposure to professional sprayers of these compounds was considerably lower. It has been shown that TCDD levels in professional applicators increase considerably above background only after several years of spraying of TCDD-contaminated chemicals. For example, in the most heavily exposed applicators of 2,4,5-T in New Zealand, who applied this chemical for at least 180 months, the estimated mean serum concentration of TCDD at the time of blood drawing was 53 ng/kg (Smith et al., 1992). Occasional spraying is unlikely to lead to any measurable increase in TCDD level.
The evaluation of the evidence of carcinogenicity of TCDD was based on studies with direct measurements of TCDD and studies involving heavy exposure to herbicides likely to be contaminated with TCDD. There are numerous studies that assessed dioxins, furans and polychlorobiphenyls (PCBs) in workers, but these are not systematically reviewed here. These studies indicate that the highest exposure occurs in industrial settings producing 2,4,5-T. There are also numerous studies in workers evaluating a wide range of health effects. Table 2.1 (available at http://monographs.iarc.fr/ENG/Monographs/vol100F/100F-22-Table2.1.pdf) shows estimated exposures to TCDD in industrial workers, in workers handling and spraying 2,4,5-T and in the population in Seveso, Italy. Average exposures in industrial workers are the highest. The effects of TCDD and those of the products in which it was found cannot be separated in most of the epidemiological studies; however, the focus here is on the contaminant. The most important studies for the evaluation of the carcinogenicity of TCDD are cohort studies of herbicide producers, one each in the United States (Fingerhut et al., 1991; Steenland et al., 1999, 2001), the Netherlands (Bueno de Mesquita et al., 1993; Hooiveld et al., 1998; Boers et al. 2010), two in Germany (Manz et al., 1991; Ott et al., 1993; Flesch-Janys et al., 1995, 1998; Becher et al., 1996), and one cohort of residents in a contaminated area from Seveso, Italy (Bertazzi et al., 2001; Pesatori et al., 2009). These studies involve the highest exposures to TCDD among all epidemiological studies, although the exposures at Seveso were lower and the follow-up was shorter than those in the industrial settings. In addition, the multicountry cohort study from IARC (Saracci et al., 1991; Kogevinas et al., 1995, 1997) is of special interest because it includes three of four high-exposure cohorts and other industrial cohorts, many of them not reported in separate publications, as well as information on professional applicators. Most of the industrial cohort studies include analyses of subcohorts considered to have the highest exposure and/or longest latency. These cohorts and their respective high-exposure subcohorts are the focus of this evaluation. In reporting the findings, preference has been given to the most updated follow-up, unless earlier publications presented evidence not included in a later publication, e.g. results by exposure classifications of interest. Results are presented in Table 2.2 (available at http://monographs.iarc.fr/ENG/Monographs/vol100F/100F-22-Table2.2.pdf), Table 2.3 (available at http://monographs.iarc.fr/ENG/Monographs/vol100F/100F-22-Table2.3.pdf), Table 2.4 (available at http://monographs.iarc.fr/ENG/Monographs/vol100F/100F-22-Table2.4.pdf), and Table 2.5 (available at http://monographs.iarc.fr/ENG/Monographs/vol100F/100F-22-Table2.5.pdf). Additional studies of herbicide applicators, both cohort and case–control studies, which have considerably lower exposures to TCDD, are not considered critical for the evaluation and are not reported in the tables. Among the studies not included, there are several that have been widely quoted and that have been important in responding to concerns in the community and in raising public awareness regarding potential effects of dioxin exposure, such as Ketchum & Aktar (1996), Ketchum et al. (1999), and Hardell & Sandström (1979).
2.1. Description of the most informative studies
2.1.1. US NIOSH cohort
The largest study of production workers in the US exposed to PCDDs was conducted by the National Institute for Occupational Safety and Health (NIOSH) and published by Fingerhut et al. (1991) and Steenland et al. (1999, 2001). This 12-plant cohort study included most workers in the USA likely to have been exposed to TCDD in chemical manufacturing, comprising 5000 men with work records showing assignment to a production or maintenance job in a process involving TCDD contamination. Serum levels of TCDD in 253 cohort members at two plants measured in 1987 averaged 233 ng/g lipid, compared with 7 ng/g lipid in a group of 79 unexposed workers. Levels increased to 418 ng/kg for 119 workers exposed for more than one year. Estimates of TCDD exposure were based on occupational records, on an exposure matrix based on industrial hygiene measurements, and on modelling based on measured TCDD in serum samples. A series of publications on two separate plants were available (Zack & Suskind, 1980; Zack & Gaffey, 1983; Cook et al., 1986; Ott et al., 1987; Bond et al., 1989; Collins et al., 1993).
2.1.2. German accident cohort
In the 1953 accident at the 2,4,5-trichlorophenol (TCP) production unit of BASF at Ludwigshafen, Germany, the total number of employees identified as being involved directly or in the subsequent clean-up, repair or maintenance activities was 247 (243 men, 4 women). Analyses of adipose tissue and blood from groups of these workers were conducted. Part of the cohort was first studied by Thiess et al. (1982) and was completed by Ott & Zober (1996).
2.1.3. IARC multicountry study
An international cohort of workers exposed to phenoxy herbicides and chlorophenols was set up by the International Agency for Research on Cancer, France (Saracci et al., 1991). The cohort included 16 863 men and 1527 women employed in production or spraying, distributed among 20 cohorts from ten countries. Exposure assessment was based on plant-production records collected in each factory through questionnaires and on detailed individual job histories. Two nested case–control studies of soft-tissue sarcoma and non-Hodgkin lymphoma were conducted by Kogevinas et al. (1995). The international cohort studied by Saracci et al. (1991) was updated and expanded with the data of Fingerhut et al. (1991), Becher et al. (1996), and Kogevinas et al. (1997). TCDD was measured in serum samples for workers in Germany, the Netherlands and the USA. Results from cohorts in specific countries have been published separately (Coggon et al., 1991; Lynge, 1993).
2.1.4. German cohorts
Several reports have considered workers from a chemical plant operated by Boehringer-Ingelheim, Hamburg, Germany. This plant produced herbicides heavily contaminated with TCDD and other PCDDs/PCDFs (Manz et al., 1991; Flesch-Janys et al., 1995, 1998; Becher et al., 1996). In the latter study, workers from three other German plants were also considered. TCDD analyses were done on serum samples from the workers in the Boehringer-Ingelheim cohort.
2.1.5. Dutch cohorts
The mortality of two cohorts of workers employed between 1955 and 1986 in the synthesis and formulation of phenoxy herbicides and chlorophenols in the Netherlands has been studied (Bueno de Mesquita et al., 1993). In one of the plants (A), where the production was focused on 2,4,5-T and derivatives, an accident in 1963 caused a release of PCDDs, including TCDD. Serum samples have been analysed for the presence of TCDD. The study has been updated (Hooiveld et al., 1998; Boers et al., 2010)
2.1.6 Seveso population exposed during an industrial accident
On 10 July 1976, a runaway reaction led to a blow-out of a TCP-production reactor at the ICMESA plant at Seveso, ltaly. The chemical cloud that was released from the reactor contained a substantial amount of TCDD. The contaminated area was subdivided into Zones A and B, and Zone R in descending order of TCDD contamination in the soil. The mortality and cancer incidence in the population of Seveso exposed during this industrial accident were investigated. The exposed and referent populations were followed-up as if they belonged to a unique cohort, blind to the exposure status of the subjects. The follow-up after 20 years was > 99% successful (Bertazzi et al., 2001; Pesatori et al., 2009)
2.2. All cancers combined
An increased risk for all cancers combined was found in the industrial cohort studies cited above in the USA, Germany, and the Netherlands and to a lesser extent in the international cohort (see Table 2.2, online). The magnitude of the increase is generally small. It is higher in subcohorts considered to have the heaviest TCDD exposure, e.g. the chloracne subcohort in the NIOSH study. Furthermore, statistically significant positive dose–response trends for all cancers combined were present in the NIOSH cohort and in the largest and most heavily exposed German cohort. A positive trend (P = 0.05) was also seen in the smaller German cohort where an accident occurred with release of large amounts of TCDD. However, the positive trend in this cohort was limited to smokers. Cumulative dose in these trend analyses was estimated by combining data from TCDD concentrations in blood and information on job categories, work processes and calendar time of exposure. A metanalysis of data from three cohorts occupationally exposed to TCDD and related compounds (NIOSH, Boehringer-Ingelheim, Germany, and BASF, Germany) found a statistically significant (P = 0.02) trend in total cancer mortality with increasing exposure to dioxin (Crump et al., 2003). The trend tests show an increase in total cancers at cumulative TEQ – a metric TCDD-like compounds that is defined here as the amount of TCDD that would produce the same toxicity as a mixture of TCDD-like compounds – serum levels that would result from lifetime intake of 7 pg TEQ/kg body weight/day. A linear dose–response provided a good fit to the combined data. There was no overall increase of cancer in the population in Seveso, with only minor increases observed for all-cancer mortality and incidence at 15 or more years since the accident in the most heavily exposed zones. [The overall increase identified in all industrial cohorts, and the positive trends with increased exposure that are based on internal comparisons, reinforce an overall positive association between all cancers combined and exposure to TCDD, making it less likely that the increase is explained by confounding, either by smoking or by other exposures to carcinogens in the industrial setting.]
2.3. Cancer of the lung
An increased risk for lung cancer was observed in the industrial cohort studies, especially in the more highly exposed subcohorts (see Table 2.3, online). The relative risk for lung cancer in the highly exposed subcohorts was around 1.5 in several studies. [It is possible that relative risks of this order for lung cancer could result from confounding by smoking, but this would only be the case if there were a pronounced difference in smoking habits between the exposed and the referent populations, a difference that seems unlikely. Therefore, confounding by smoking can probably not explain all the excess risk for lung cancer, although it could explain part of it. It is also possible that other occupational carcinogens, many of which would affect the lung, are causing some confounding.] In Seveso, increased mortality and cancer incidence for lung cancer was observed at more than 15 years since the accident.
2.4. Soft-tissue sarcoma
An association between soft-tissue sarcoma and spraying of phenoxy herbicides was first suggested by results from case–control studies in Umea, Sweden (Hardell & Sandström, 1979). Exposure to TCDD in these and other community-based case–control studies is, however, not accurately estimated. An excess risk for soft-tissue sarcoma, based on a small number of deaths, has been reported in the largest industrial cohorts, specifically those of NIOSH and IARC (see Table 2.4, online). In both, the mortality ratios (SMRs) tended to be higher among the most exposed subcohorts. Incidence data for soft-tissue sarcoma were generally not available. A dose–response relationship, with estimated exposure to TCDD, was found in a case–control study nested in the IARC cohort; however, strong positive trends were also found with exposure estimates for 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T). In Seveso, there were no cases of soft-tissue sarcoma in the most heavily contaminated Zones A and B. [Soft-tissue sarcomas are subject to serious misclassification on death certificates. Although it is unlikely that this occurs differentially in the exposed and the referent populations, re-classification of a few cases would have important consequences on results based on small numbers.]
2.5. Non-Hodgkin lymphoma
An increased risk for non-Hodgkin lymphoma was found in most of the populations studied in the four industrial cohort studies and in the Seveso population, although the relative risks were mostly non-significant and below 2 (see Table 2.5, online). A case–control study nested in the IARC cohort provided weak evidence of a dose–response relationship with estimated exposure to TCDD. [Although it is plausible that other chemicals cause non-Hodgkin lymphoma, strong potential confounding factors are not known. The lack of complete consistency among the cohorts and the weak effect detected in most of the positive studies, however, caution against a causal interpretation of the findings.]
2.6. Other cancers
Increased risks for several other malignant neoplasms have been sporadically reported among workers exposed to TCDD, and at Seveso. Most notable are risks for breast and rectal cancers and myeloid leukaemia in Seveso, bladder cancer in the NIOSH and Dutch cohorts, multiple myeloma in the NIOSH cohort, cancers of the oral cavity and pharynx in the German cohorts, genital cancers in the Dutch cohort, and kidney cancer in the IARC cohort. [The available results are not fully consistent, and several studies have not reported the results for each individual cancer site.]
2.7. Synthesis
Overall, the strongest evidence for the carcinogenicity of TCDD is for all cancers combined, rather than for any specific site. The relative risk for all cancers combined in the most highly exposed and longer-latency subcohorts is around 1.4. In dose–response analyses, higher relative risks are observed for the groups with the highest measured and modelled exposure to TCDD. This relative risk for all neoplasms does not appear likely to be explained by confounding, particularly since dose–response was typically based on internal comparisons among workers of the same cohort. The evidence for specific cancers is strongest for lung cancer, soft-tissue sarcoma and non-Hodgkin lymphoma, but confounding cannot be ruled out for lung cancer, while the findings on soft-tissue sarcoma are based on small numbers. Several studies identified statistically significant increases in many cancers, but findings for other cancers including major cancers are, overall, inconsistent between studies. It should be borne in mind that the general population is exposed to levels that are much lower than those experienced by the industrial populations.
The Working Group did not review the epidemiological evidence of other PCDDs, PCDFs or PCBs with a dioxin-like activity.
3. Cancer in Experimental Animals
3.1 2,3,7,8-Tetrachlorodibenzo-para-dioxin
Carcinogenicity studies with several strains of rats, mice and Syrian hamsters treated with 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) via the oral route (gavage or diet), by intra-peritoneal injection, or by skin application have been reviewed in IARC Monograph Volume 69 (IARC, 1997). At the time, the review of the available data led to the conclusion that there is sufficient evidence in experimental animals for the carcinogenicity of TCDD. The present Monograph also evaluates relevant carcinogenicity studies in TCDD-treated experimental animals that were published since 1997. The results of adequately conducted carcinogenicity studies are summarized below and in Table 3.1 and Table 3.2.
Table 3.1
Carcinogenicity studies in mice exposed to 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD).
Table 3.2
Carcinogenicity studies in rats and hamsters exposed to 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD).
TCDD was tested for carcinogenicity by oral administration (gavage or dose feed) in four studies in mice and six studies in rats, by skin (topical) application in two studies in mice, by intraperitoneal injection in one study in mice, one study in rats and one study in hamsters and by subcutaneous injection in one study in hamsters. TCDD produced tumours in both sexes of mice and rats, and in multiple organs and tissues.
Oral administration of TCDD caused increased incidences of thyroid follicular adenomas and hepatocellular adenomas and carcinomas in male and female mice, of alveolar/bronchiolar adenomas and carcinomas in male mice, and of histiocytic lymphomas and subcutaneous fibrosarcomas in female mice. In rats, it caused increased incidences of hepatocellular adenomas in males and females, cholangiocarcinomas and hepatocellular carcinomas in females, lung cystic keratinizing epitheliomas and squamous-cell carcinomas in females, adrenal gland (cortex) adenomas and squamous-cell carcinomas of the hard palate/nasal turbinates in males and females, tongue squamous-cell carcinomas and thyroid follicular adenomas and carcinomas combined in males, subcutaneous fibromas in males and subcutaneous fibrosarcomas in females, and pituitary adenomas, uterine and oral mucosa (gingival) squamous-cell carcinomas and pancreatic adenomas and carcinomas combined in females (Van Miller et al., 1977; Kociba et al., 1978; Tóth et al., 1979, NTP, 1982a, 2006a; Della Porta et al., 1987; Goodman & Sauer, 1992; Hays et al., 1997, Yoshizawa et al., 2005). Skin application or gavage caused benign and malignant tumours of the skin in female mice including transgenic mice (NTP, 1982b; Wyde et al., 2004). Hamsters that received TCDD by intraperitoneal or subcutaneous injection developed squamous-cell carcinomas of the facial skin (Rao et al., 1988). Intraperitoneal injection caused increased incidence of hepatocellular adenomas and carcinomas in female mice and of lymphomas in male and female mice (Della Porta et al., 1987).
Several studies in mice showed that administration of TCDD with known carcinogens enhanced the incidence of skin papillomas, lung adenomas, liver adenomas and hepatoblastomas. In female rats, TCDD co-administered with various nitrosamines enhanced the incidence of focal hepatic lesions. In one study, TCDD enhanced the incidence of lung carcinomas in ovariectomized female rats following administration of N-nitrosodiethylamine (NDEA) (IARC, 1997). In two more recent studies in female rats, TCDD given orally or subcutaneously enhanced the carcinogenicity of previously administered NDEA (Davis et al., 2000; Viluksela et al., 2000). In another study, the oral administration of TCDD to pregnant rats increased 7,12-dimethylbenz[a]anthracene-induced mammary-gland tumours in offspring (Brown et al., 1998; see Table 3.3).
Table 3.3
Studies on 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) administered to rats, in combination with known carcinogens or modifying factors.
3.2. Dioxin-like compounds
3.2.1. 2,3,4,7,8-Pentachlorodibenzofuran
Oral administration of 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) resulted in significant dose-dependent trends for increased incidence of cholangiocarcinomas and hepatocellular adenomas (Walker et al., 2005; NTP, 2006b). (see Table 3.4)
Table 3.4
Carcinogenicity and initiation-promotion studies in experimental animals exposed to 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) and 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126).
Skin application of PeCDF after a single dose of N-methyl-N′-nitro-N-nitrosoguanidine resulted in an increased incidence of skin papillomas in mice (Hébert et al., 1990). Subcutaneous injections of PeCDF after oral treatment with NDEA resulted in an increased multiplicity of hepatocellular carcinomas and liver hyperplastic nodules in male rats (Nishizumi & Masuda, 1986). Subcutaneous injections of PeCDF after a single intraperitoneal injection of NDEA increased the number of focal hepatic lesions in female rats (Waern et al., 1991).
3.2.2. 3,3′,4,4′,5-Pentachlorobiphenyl
Oral administration of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) resulted in significantly increased incidence of hepatocellular adenomas, cholangiocarcinomas, lung cystic keratinizing epitheliomas, and oral mucosa (gingiva) squamous-cell carcinomas in female rats (Walker et al., 2005; NTP, 2006c).
4. Other Relevant Data
4.1. AhR activation
Most, if not all of the effects of TCDD are related to its binding to and activation of the aryl hydrocarbon receptor (AhR), a member of the basic helix–loop–helix/Per-Arnt-Sim family of transcription factors. This receptor was first identified in mouse liver (Poland et al., 1976) where it showed high affinity towards TCDD. Further studies found that AhR is expressed in most mammalian tissues and that many other halogenated aromatic compounds can bind this receptor, including the coplanar polychlorinated biphenyls and the polychlorinated dibenzodioxins and dibenzofurans. It is generally proposed that the toxic and carcinogenic effects of dioxin and other halogenated compounds are due to their high affinity to AhR, and to the sustained pleiotropic response from a battery of genes – many of which encode drug-metabolizing enzymes – that follows the receptor-ligand complex formation (Mandal, 2005; Walker, 2007). Much of the research in the three decades since the discovery of the AhR has focused on dissecting this pleiotropic response to fully understand the mechanisms involved in dioxin-mediated toxicity.
The free AhR resides in the cytoplasm as an inactive complex containing a heat-shock protein dimer, Hsp90, XAP2 and p23 (Meyer et al., 1998). When the AhR binds to a ligand, XAP2 is released and, through a conformational change, the complex is moved to the nucleus where the Hsp90 dimer dissociates and the AhR-nuclear-translocator (ARNT) binds to the PAS domains of the receptor. The activated AhR/ARNT complex forms a heterodimer that is then capable of binding to the 5′-regulatory region of dioxin-responsive genes (Mimura & Fujii-Kuriyama, 2003). The primary targets following activation of AhR include genes encoding many phase-I and phase-II metabolic enzymes (e.g. CYP1A1, CYP1A2, CYP1B1, NQO1, UGT1A2, GSTA1 and ALDH3A1) (Nebert et al., 2000; Schwarz & Appel, 2005). However, through direct and indirect pathways, TCDD is able to alter the expressions of a much larger number of genes (Martinez et al., 2002; Dere et al., 2006; Pastorelli et al., 2006; Schwanekamp et al., 2006). In addition, there is cross-talk with several other receptor-mediated systems including the estrogen receptor (Safe & Wormke, 2003) and the retinoic-acid receptor β (Lu et al., 1994; Berkers et al., 1995; Toyoshiba et al., 2004).
Despite the strong conservation of the AhR across species, gene polymorphisms, differences in co-activators and differences in downstream signalling following activation are all likely to modulate TCDD carcinogenicity (Ema et al., 1994; Tuomisto et al., 1999). These factors could explain the interindividual differences observed in the magnitude of the carcinogenic response after exposure to TCDD. For example, different AhR polymorphisms triggered a threefold difference in EROD activity in human lymphocytes (Smart & Daly, 2000).
4.2. Mechanisms of carcinogenicity
TCDD is not directly genotoxic and the tumorigenic activity is likely to be due to a fairly long half-life, especially in humans, resulting in a sustained activation of the AhR. TCDD half-life in the human body is estimated at 7.2 years (Milbrath et al., 2009); long half-life in the environment and the ability to bio-accumulate in the food-chain are also reported (IARC, 1997). The sustained downstream signalling may trigger an adaptive biochemical and physiological response in the cell that can promote carcinogenesis (Biegel & Safe, 1990; Lu et al., 1994; Berkers et al., 1995; Schwarz & Appel, 2005), also by inducing mutations (Stohs et al., 1990; Tritscher et al., 1996; Shertzer et al., 1998; Yoshida & Ogawa, 2000; Thornton et al., 2001; Nebert et al., 2004; Knerr et al., 2006; Schlezinger et al., 2006; Lin et al., 2007; Green et al., 2008). TCDD may also enhance – although it sometimes inhibits – the progression and invasiveness of initiated tumours (Marlowe & Puga, 2005; Peng et al., 2009), but this topic will not be discussed in detail here.
The primary mechanism by which TCDD is thought to cause cancer is by altering the cellular ability to proliferate, migrate, apoptose, senesce and terminally differentiate (Safe, 2001; Marlowe & Puga, 2005; Ray & Swanson, 2009) in a multistep process focused on the accumulation of mutations and/or heritable epigenetic changes. Chemicals that inhibit apoptosis and increase proliferation usually increase cancer risk as well. TCDD has been shown to increase cellular proliferation both in vivo and in vitro in several tissues (Maronpot et al., 1993; Barrett, 1995; Dere et al., 2006) possibly through interactions with protein-kinase C signalling (Barrett, 1995), inhibition of senescence (Ray & Swanson, 2009) or activation of growth-signalling factors (Kohn, 1995). In initiation-promotion models, TCDD expanded the populations of preneoplastic foci in rat liver (Dragan et al., 1992; Maronpot et al., 1993; Tritscher et al., 1995) and promoted carcinogenesis in liver, skin and lung in rodents (DiGiovanni et al., 1977; Hébert et al., 1990; Lucier et al., 1991; Dragan et al., 1992; Beebe et al., 1995; Tritscher et al., 1995; Tritscher et al., 2000).
Finally, TCDD may upregulate drug-metabolizing enzymes, thus increasing the presence of highly reactive intermediates that form during metabolic activation and/or transformation of several key hormones. For example, CYP1A1, CYP1A2 and CYP1B1 induction is a major source of reactive oxygen species (ROS) formation in hepatocytes and this has been linked to the decoupling of the P450 catalytic cycle (Nebert et al., 2004; Knerr et al., 2006; Schlezinger et al., 2006; Green et al., 2008). A hormonal linkage with estrogen has been demonstrated through the increase in 8-oxo-deoxyguanosine (a marker of oxidative stress) in the liver of intact female rats compared with rats that are ovariectomized before exposure to TCDD (Tritscher et al., 1996). TCDD may induce prolonged oxidative stress, and consequent DNA damage and mutations, also in several other strains of rats and mice, and in cell lines (Stohs et al., 1990; Shertzer et al., 1998; Yoshida & Ogawa, 2000; Thornton et al., 2001; Wyde et al., 2001; Lin et al., 2007). TCDD is a complete carcinogen in mice and rats in multiple strains (IARC, 1997; NTP, 2006a); see Section 3).
TCDD thus may both promote and initiate carcinogenesis through indirect oxidative stress, leading some to refer to dioxin as an activator of carcinogenesis and to adopt initiation-promotion models to better explain the toxicity of TCDD and better fit pre-neoplastic and neoplastic data (Portier & Kohn, 1996; Portier et al., 1996; Luebeck et al., 2000).
4.3. Dioxin-like compounds
As noted above, the carcinogenicity of TCDD is tied to a sustained, pleiotropic response following exposure to dioxin. The 17 laterally-substituted (2,3,7,8-substituted) polychlorinated dibenzodioxins (PCDD-Ls), the coplanar polychlorinated biphenyls (CP-PCBs) and the 17 laterally-substituted polychlorinated dibenzofurans (PCDF-Ls) are all structurally and toxicologically inter-related halogenated aromatic hydrocarbons referred to as dioxin-like compounds (DLCs). They potentially induce pleiotropic responses in cells very similar to those induced by TCDD (IARC, 1978, 1987, 1997; Vezina et al., 2004) as they are all able to bind to the AhR. Binding affinity is different among DLCs, with some of them having so little binding affinity that there is almost no information on their biological impact. We focus here on the 28 DLCs (DLC-28) listed in Table 4.1 that produce a TCDD-like response in human cell lines (Endo et al., 2003), in animal and primary human cells, and in animal and human tissue (Vezina et al., 2004; Silkworth et al., 2005; Kopec et al., 2008; N’Jai et al., 2008).
Table 4.1
Toxicity Equivalence Factors (TEFs) and half-lives of the dioxin-like compounds.
DLCs co-occur in virtually every environmental compartment where they are found, and assessment of their potential effect on human risk can be complicated. Public health authorities (in particular WHO expert panels) have developed the toxicity equivalence factor (TEF) methodology for measuring the potency of DLCs, with TCDD as the index chemical. Exposures are calculated as a simple weighted sum of the individual amounts multiplied by their individual TEFs to yield the equivalent dose in units of TCDD exposure. Only the compounds of the DLC-28 series have TEF values > 0 (Table 4.1), all of the remaining congeners have TEF = 0.
WHO recently evaluated TEFs (Van den Berg et al., 2006) thus reviewing much of the literature on DLCs (Haws et al., 2006), namely 189 studies with over 1000 targets used for analysis. For every compound, there is at least one in vitro study showing AhR binding and one showing 7-ethoxyresorufin-O-dethylase activity, which is associated with an increase in CYP1A1 activity. Congeners 1,2,3,7,8-PeCDD, 2,3,7,8-TCDF, 2,3,4,7,8-PeCDF, and PCB-77, -126, and -169 have a very broad spectrum of information both in animal and in human cells, demonstrating activity consistent with the mechanisms described earlier. In addition, for PCB-81 there are nine studies in human cell lines showing the same alterations in gene expression and enzyme activity as reported for TCDD.
Congeners 1,2,3,7,8-pentaCDD, 1,2,3,4,6,7,8-heptaCDD, a mixture of 1,2,3,6,7,8- and 1,2,3,7,8,9-hexaCDD, and a mixture of PCDDs have been studied in two-stage experimental models (IARC, 1978) and shown to enhance the carcinogenic potential of known carcinogens just like TCDD. There are some human studies on the compounds in the DLC-28 series, but they have either poor exposure characterization or also contain TCDD, making it difficult to interpret their individual effects (IARC, 1978).
While the carcinogenicity of TCDD has been clearly established in rodents, for the remaining compounds in the DLC-28 series bioassays on two-year chronic exposure are lacking. A two-year chronic study on 2,3,4,7,8-PeCDF in rodents (NTP, 2006b) demonstrated tumour effects consistent with those seen for TCDD (hepatocellular adenomas, cholangiocarcinomas, gingival squamous cell carcinomas, and an equivocal finding of lung cystic keratonizing epitheliomas). When compared for potency, the result from this study agreed with the TEF concept (Walker et al., 2005). 2,3,4,7,8-PeCDF and 1,2,3,4,7,8-HxCDF were also enhanced tumorigenesis in two-stage studies of cancer (IARC, 1997). Follow-up of populations in Taiwan, China (Tsai et al., 2007) and Japan (Onozuka et al., 2009) accidentally exposed to rice-oil containing PCDFs and PCBs, shows a significantly increased risk of mortality from chronic liver disease in men and a non-significant increase from liver cancer in men and women in Taiwan, China while in Japan all cancer mortality, and liver and lung cancer-mortalities were increased in men.
The carcinogenicity of mixtures of PCBs in rodents has also been clearly established through studies of various Aroclors (IARC, 1978; Mayes et al., 1998; NTP, 2006c) yielding predominantly liver cancers (Cogliano, 1998). Two-year chronic exposure studies done by the US National Toxicology Program (NTP) on PCB 126 (NTP, 2006d) and PCB 118 (NTP, 2009), demonstrated tumour effects consistent with those seen for TCDD (hepatocellular adenomas, cholangiocarcinomas, gingival squamous cell carcinomas, and lung cystic keratonizing epitheliomas). Moreover, when equivalent TCDD doses where applied with the current TEF, a carcinogenic response equivalent to that predicted for TCDD from the NTP study (Walker et al., 2005) was observed.
The set of DLC-28 (IARC, 1978, 1997; Milbrath et al., 2009) have a long half-life similar to that of TCDD (estimated at 7.2 years in the human body) (Table 4.1). Many congeners have similar or longer half-lives (1,2,3,7,8-PeCDD, 1,2,3,4,7,8- and 1,2,3,6,7,8-HxCDD, 1,2,3,6,7,8- and 1.2.3.7.8.9-HxCDF, and PCBs 169, 114, 123, 156, 167 and 189) while most of the remaining half-lives are in excess of 1.4 years. Several authors report the presence of these compounds in human blood in the general population (Costopoulou et al., 2006; Scott et al., 2008; Zubero et al., 2009) indicating a sustained, long-term exposure that, when coupled with the analyses for common pleiotropic response, argues in favour of the notion that all of the DLC-28s have the same carcinogenic potential in humans.
Experimental data on mechanism of carcinogenesis induced by DLC-28 are available for 2,3,4,7,8-PeCDF and PCB 126, in particular (Table 4.2), Both have been shown to bind to the AhR in humans and animals (IARC, 1978; Safe, 2001), to translocate into the nucleus and activate numerous metabolic enzymes in vitro (human and non-human cell lines) and in vivo in experimental animals (IARC, 1997; Safe, 2001; Vezina et al., 2004; Haws et al., 2006), to trigger changes in growth factors and signalling pathways related to cellular replication in rodents (Hemming et al., 1995; Vondrácek et al., 2005; N’Jai et al., 2008). 2,3,4,7,8-PeCDF potential effect on cell replication is suggested in the NTP study (Walker et al., 2007), and promotion in skin, liver and lung tissues is reported in initiation-promotion studies (Hébert et al., 1990; Anderson et al., 1991; Waern et al., 1991). PCB 126 acts as a promoter of liver cancer in initiation-promotion studies (Hemming et al., 1995; Haag-Grönlund et al., 1998) with measured increases in cell-replication rate in the populations of initiated cells (Vondrácek et al., 2005). PCB 126 and 2,3,4,7,8-PeCDF induce oxidative stress, the latter in a dose-dependent manner in brain and liver of rats (Hassoun et al., 2002; Hennig et al., 2002). These two compounds are carcinogenic in mixtures with TCDD (IARC, 1978; Hassoun et al., 2001; NTP, 2006d) and by themselves in the NTP chronic bioassays in rats, where they increase hepatocellular adenomas, cholangiocarcinomas, gingival squamous-cell carcinomas, and, possibly, lung cystic keratonizing epitheliomas (NTP, 2006b, c, d).
Table 4.2
Experimental evidence on the mechanisms of carcinogenesis for TCDD and the dioxin-like compounds 2,3,4,7,8-PeCDF and PCB 126 (positive, non-determined, and indirect).
4.4. Synthesis
There is strong evidence to support a receptor-mediated mechanism of action for TCDD-associated carcinogenesis in humans where the primary mechanism is the promotion of tumour development through the activation of cellular replication and the alteration in cellular senescence and apoptosis. Dioxin, through activation of an array of metabolic enzymes also increases the risk for oxidative stress, which serves as an indirect initiator of carcinogenesis. These events make dioxin a complete carcinogen. The conservation of the AhR and the related signalling pathways across species strongly support this mechanism in humans.
The receptor-mediated mechanism of action for TCDD-associated carcinogenesis in humans is strongly suggested as the mechanism of action that would result in 2,3,4,7,8-PeCDF and PCB 126 causing cancer in humans. The primary mechanism is the promotion of carcinogenesis through the activation of cellular replication and the alteration in cellular senescence and apoptosis through the aryl-hydrocarbon receptor (AhR). These congeners, through activation of an array of metabolic enzymes, increase the risk for oxidative stress as an indirect initiator of carcinogenesis, which makes these congeners complete carcinogens. The conservation of the AhR and the related signalling pathways across species strongly support this mechanism of action in humans.
There is compelling evidence that the mechanism of action for TCDD-associated carcinogenesis in humans operates as the mechanism of action for carcinogenesis in humans for 1,2,3,7,8-PeCDD, 1,2,3,4,7,8-HxCDD, 1,2,3,6,7,8-HxCDD, 1,2,3,7,8,9-HxCDD, 1,2,3,4,6,7,8-HpCDD, OCDD, 2,3,7,8-TCDF, 1,2,3,7,8-PeCDF, 1,2,3,4,7,8-HxCDF, 1,2,3,6,7,8-HxCDF, 1,2,3,7,8,9-HxCDF, 2,3,4,6,7,8-HxCDF, 1,2,3,4,6,7,8-HpCDF, 1,2,3,4,7,8,9-HpCDF, OCDF and PCBs 77, 81, 105, 114, 118, 123, 156, 157, 167, 169, and 189. These compounds all bind to the AhR in human cells and demonstrate changes in gene expression consistent with those seen for TCDD, 2,3,4,7,8-PeCDF and PCB 126. Where examined, data have been collected for these compounds supporting some, but not all, aspects of the mechanisms outlined for TCDD, 2,3,4,7,8-PeCDF and PCB 126 that relate to activation of cell replication, alterations in cellular senescence and apoptosis, and increases in oxidative stress.
5. Evaluation
There is sufficient evidence in humans for the carcinogenicity of 2,3,7,8-tetrachlorodibenzo-para-dioxin. The strongest evidence in humans for the carcinogenicity of 2,3,7,8-tetrachlorodibenzo-para-dioxin is for all cancers combined.
Also, a positive association has been observed between exposure to 2,3,7,8-tetrachlorodibenzo-para-dioxin and soft-tissue sarcoma, non-Hodgkin lymphoma and cancer of the lung.
There is sufficient evidence in experimental animals for the carcinogenicity of 2,3,7,8-tetrachlorodibenzo-para-dioxin.
There is sufficient evidence in experimental animals for the carcinogenicity of 2,3,4,7,8-pentachlorodibenzofuran.
There is sufficient evidence in experimental animals for the carcinogenicity of 3,3′,4,4′,5-pentachlorobiphenyl.
There is strong evidence to support a receptor-mediated mechanism that operates in humans for carcinogenesis associated with 2,3,7,8-tetrachlorodibenzo-para-dioxin, where the primary mechanism is the promotion of tumour development through modification of cell replication and apoptosis, with a secondary mechanism related to increases of oxidative stress causing DNA damage. The conservation of the aryl hydrocarbon receptor and the related signalling pathways and responses across species, including humans, add additional strength to the notion that this mechanism is active in humans.
2,3,7,8-Tetrachlorodibenzo-para-dioxin is carcinogenic to humans (Group 1).
2,3,4,7,8-Pentachlorodibenzofuran is carcinogenic to humans (Group 1).
3,3′,4,4′,5-Pentachlorobiphenyl is carcinogenic to humans (Group 1).
In making the second and third overall evaluations, the Working Group considered the following mechanistic arguments:
There is strong evidence to support a receptor-mediated mechanism for 2,3,4,7,8-pentachlorodibenzofuran- and 3,3′,4,4′,5-pentachlorobiphenyl-associated carcinogenesis in humans based upon evidence of carcinogenicity in experimental animals and upon extensive evidence showing activity identical to 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) for every step of the mechanism described for TCDD-associated carcinogenesis in humans including receptor binding, gene expression, protein-activity changes, cellular replication, oxidative stress, promotion in initiation-promotion studies and complete carcinogenesis in laboratory animals.
References
- Anderson LM, Beebe LE, Fox SD, et al. Promotion of mouse lung tumors by bioaccumulated polychlorinated aromatic hydrocarbons. Exp Lung Res. 1991;17:455–471. [PubMed: 1904809]
- ATSDR (1998). Toxicological Profile for Chlorinated Dibenzo-p-dioxins. Atlanta, GA, pp. 21.
- Aylward LL, Bodner KM, Collins JJ, et al. TCDD exposure estimation for workers at a New Zealand 2,4,5-T manufacturing facility based on serum sampling data. J Expo Sci Environ Epidemiol. 2010;20:417–426. [PubMed: 19491942]
- Aylward LL, Hays SM. Temporal trends in human TCDD body burden: Decreases over three decades and implications for exposure levels. J Expo Anal Environ Epidemiol. 2002;12:319–328. [PubMed: 12198580] [CrossRef]
- Baars AJ, Bakker MI, Baumann RA, et al. Dioxins, dioxin-like PCBs and non-dioxin-like PCBs in foodstuffs: occurrence and dietary intake in The Netherlands. Toxicol Lett. 2004;151:51–61. [PubMed: 15177640] [CrossRef]
- Baccarelli A, Mocarelli P, Patterson DG Jr, et al. Immunologic effects of dioxin: new results from Seveso and comparison with other studies. Environ Health Perspect. 2002;110:1169–1173. [PMC free article: PMC1241102] [PubMed: 12460794]
- Barrett JC. Mechanisms for species differences in receptor-mediated carcinogenesis. Mutat Res. 1995;333:189–202. [PubMed: 8538627]
- Bates MN, Buckland SJ, Garrett N, et al. Persistent organochlorines in the serum of the non-occupationally exposed New Zealand population. Chemosphere. 2004;54:1431–1443. [PubMed: 14659945] [CrossRef]
- Becher H, Flesch-Janys D, Kauppinen T, et al. Cancer mortality in German male workers exposed to phenoxy herbicides and dioxins. Cancer Causes Control. 1996;7:312–321. [PubMed: 8734824] [CrossRef]
- Becher H, Steindorf K, Flesch-Janys D. Quantitative cancer risk assessment for dioxins using an occupational cohort. Environ Health Perspect. 1998;106 Suppl 2):663–670. [PMC free article: PMC1533398] [PubMed: 9599714] [CrossRef]
- Beebe LE, Fornwald LW, Diwan BA, et al. Promotion of N-nitrosodiethylamine-initiated hepatocellular tumors and hepatoblastomas by 2,3,7,8-tetrachlorodibenzo-p-dioxin or Aroclor 1254 in C57BL/6, DBA/2, and B6D2F1 mice. Cancer Res. 1995;55:4875–4880. [PubMed: 7585523]
- Berkers JA, Hassing I, Spenkelink B, et al. Interactive effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and retinoids on proliferation and differentiation in cultured human keratinocytes: quantification of cross-linked envelope formation. Arch Toxicol. 1995;69:368–378. [PubMed: 7495374]
- Bertazzi PA, Consonni D, Bachetti S, et al. Health effects of dioxin exposure: a 20-year mortality study. Am J Epidemiol. 2001;153:1031–1044. [PubMed: 11390319] [CrossRef]
- Biegel L, Safe S. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on cell growth and the secretion of the estrogen-induced 34-, 52- and 160-kDa proteins in human breast cancer cells. J Steroid Biochem Mol Biol. 1990;37:725–732. [PubMed: 2278856]
- Boers D, Portengen L, Bueno-de-Mesquita HB, et al. Cause-specific mortality of Dutch chlorophenoxy herbicide manufacturing workers. Occup Environ Med. 2010;67:24–31. [PubMed: 19736176] [CrossRef]
- Bond GG, McLaren EA, Lipps TE, Cook RR. Update of mortality among chemical workers with potential exposure to the higher chlorinated dioxins. J Occup Med. 1989;31:121–123. [PubMed: 2709162]
- Brown NM, Manzolillo PA, Zhang J-X, et al. Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis. 1998;19:1623–1629. [PubMed: 9771934] [CrossRef]
- Bueno de Mesquita HB, Doornbos G, Van der Kuip DAM, et al. Occupational exposure to phenoxy herbicides and chlorophenols and cancer mortality in The Netherlands. Am J Ind Med. 1993;23:289–300. [PubMed: 8427257] [CrossRef]
- Burns CJ, Collins JJ, Budinsky RA, et al. Factors related to dioxin and furan body levels among Michigan workers. Environ Res. 2008;106:250–256. [PubMed: 18054905] [CrossRef]
- Charnley G, Doull J. Human exposure to dioxins from food, 1999–2002. Food Chem Toxicol. 2005;43:671–679. [PubMed: 15778006] [CrossRef]
- Charnley G, Kimbrough RD. Overview of exposure, toxicity, and risks to children from current levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds in the USA. Food Chem Toxicol. 2006;44:601–615. [PubMed: 16176855] [CrossRef]
- Coggon D, Pannett B, Winter P. Mortality and incidence of cancer at four factories making phenoxy herbicides. Br J Ind Med. 1991;48:173–178. [PMC free article: PMC1035345] [PubMed: 2015208]
- Cogliano VJ. Assessing the cancer risk from environmental PCBs. Environ Health Perspect. 1998;106:317–323. [PMC free article: PMC1532993] [PubMed: 9618347]
- Collins JJ, Bodner K, Haidar S, et al. Chlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyl profiles of workers with trichlorophenol and pentachlorophenol exposures. Chemosphere. 2008;73:S284–S289. [PubMed: 18442847] [CrossRef]
- Collins JJ, Bodner KM, Wilken M, et al. Serum concentrations of chlorinated dibenzo-p-dioxins and dibenzofurans among former Michigan trichlorophenol and pentachlorophenol workers. J Expo Sci Environ Epidemiol. 2007;17:541–548. [PubMed: 17426737] [CrossRef]
- Collins JJ, Budinsky RA, Burns CJ, et al. Serum dioxin levels in former chlorophenol workers. J Expo Sci Environ Epidemiol. 2006;16:76–84. [PubMed: 16015278] [CrossRef]
- Collins JJ, Strauss ME, Levinskas GJ, Conner PR. The mortality experience of workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin in a trichlorophenol process accident. Epidemiology. 1993;4:7–13. [PubMed: 8420584]
- Cook RR, Bond GG, Olson RA, et al. Evaluation of the mortalty experience of workers exposed to the chlorinated dioxins. Chemosphere. 1986;15:1769–1776. [CrossRef]
- Costopoulou D, Vassiliadou I, Papadopoulos A, et al. Levels of dioxins, furans and PCBs in human serum and milk of people living in Greece. Chemosphere. 2006;65:1462–1469. [PubMed: 16765419]
- Crump KS, Canady R, Kogevinas M. Meta-analysis of dioxin cancer dose response for three occupational cohorts. Environ Health Perspect. 2003;111:681–687. [PMC free article: PMC1241475] [PubMed: 12727594]
- Davis BJ, McCurdy EA, Miller BD, et al. Ovarian tumors in rats induced by chronic 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Cancer Res. 2000;60:5414–5419. [PubMed: 11034082]
- Della Porta G, Dragani TA, Sozzi G. Carcinogenic effects of infantile and long-term 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment in the mouse. Tumori. 1987;73:99–107. [PubMed: 3576718]
- Dere E, Boverhof DR, Burgoon LD, Zacharewski TR. In vivo-in vitro toxicogenomic comparison of TCDD-elicited gene expression in Hepa1c1c7 mouse hepatoma cells and C57BL/6 hepatic tissue. BMC Genomics. 2006;7:80. [PMC free article: PMC1513214] [PubMed: 16611356]
- DiGiovanni J, Viaje A, Berry DL, et al. Tumor-initiating ability of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and Arochlor 1254 in the two-stage system of mouse skin carcinogenesis. Bull Environ Contam Toxicol. 1977;18:552–557. [PubMed: 412534]
- Dragan YP, Xu XH, Goldsworthy TL, et al. Characterization of the promotion of altered hepatic foci by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the female rat. Carcinogenesis. 1992;13:1389–1395. [PubMed: 1354083]
- Dwernychuk LW, Cau HD, Hatfield CT, et al. Dioxin reservoirs in southern Viet Nam – a legacy of Agent Orange. Chemosphere. 2002;47:117–137. [PubMed: 11993628] [CrossRef]
- Ema M, Ohe N, Suzuki M, et al. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J Biol Chem. 1994;269:27337–27343. [PubMed: 7961644]
- Endo F, Monsees TK, Akaza H, et al. Effects of single non-ortho, mono-ortho, and di-ortho chlorinated biphenyls on cell functions and proliferation of the human prostatic carcinoma cell line, LNCaP. Reprod Toxicol. 2003;17:229–236. [PubMed: 12642156]
- Fingerhut MA, Halperin WE, Marlow DA, et al. Cancer mortality in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. N Engl J Med. 1991;324:212–218. [PubMed: 1985242]
- Flesch-Janys D, Berger J, Gurn P, et al. Exposure to polychlorinated dioxins and furans (PCDD/F) and mortality in a cohort of workers from a herbicide-producing plant in Hamburg, Federal Republic of Germany. Am J Epidemiol. 1995;142:1165–1175. [PubMed: 7485063]
- Flesch-Janys D, Steindorf K, Gurn P, Becher H. Estimation of the cumulated exposure to polychlorinated dibenzo-p-dioxins/furans and standardized mortality ratio analysis of cancer mortality by dose in an occupationally exposed cohort. Environ Health Perspect. 1998;106 Suppl 2:655–662. [PMC free article: PMC1533379] [PubMed: 9599713] [CrossRef]
- Gies A, Neumeier G, Rappolder M, Konietzka R. Risk assessment of dioxins and dioxin-like PCBs in food – comments by the German Federal Environmental Agency. Chemosphere. 2007;67:S344–S349. [PubMed: 17223171] [CrossRef]
- Goodman DG, Sauer RM. Hepatotoxicity and carcinogenicity in female Sprague-Dawley rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): a pathology working group reevaluation. Regulatory Toxicology and Pharmacology. 1992;15:245–252. [PubMed: 1509118] [CrossRef]
- Green RM, Hodges NJ, Chipman JK, et al. Reactive oxygen species from the uncoupling of human cytochrome P450 1B1 may contribute to the carcinogenicity of dioxin-like polychlorinated biphenyls. Mutagenesis. 2008;23:457–463. [PubMed: 18583386]
- Haag-Grönlund M, Johansson N, Fransson-Steen R, et al. Interactive effects of three structurally different polychlorinated biphenyls in a rat liver tumor promotion bioassay. Toxicol Appl Pharmacol. 1998;152:153–165. [PubMed: 9772211]
- Hardell L, Sandström A. Case-control study: soft-tissue sarcomas and exposure to phenoxyacetic acids or chlorophenols. Br J Cancer. 1979;39:711–717. [PMC free article: PMC2009993] [PubMed: 444410]
- Hassoun EA, Li F, Abushaban A, Stohs SJ. Production of superoxide anion, lipid peroxidation and DNA damage in the hepatic and brain tissues of rats after subchronic exposure to mixtures of TCDD and its congeners. J Appl Toxicol. 2001;21:211–219. [PubMed: 11404832]
- Hassoun EA, Wang H, Abushaban A, Stohs SJ. Induction of oxidative stress in the tissues of rats after chronic exposure to TCDD, 2,3,4,7,8-pentachlorodibenzofuran, and 3,3′,4,4′,5-pentachlorobiphenyl. J Toxicol Environ Health A. 2002;65:825–842. [PubMed: 12079609]
- Haws LC, Su SH, Harris M, et al. Development of a refined database of mammalian relative potency estimates for dioxin-like compounds. Toxicol Sci. 2006;89:4–30. [PubMed: 16120753]
- Hays SM, Aylward LL. Dioxin risks in perspective: past, present, and future. Regul Toxicol Pharmacol. 2003;37:202–217. [PubMed: 12726754] [CrossRef]
- Hays SM, Aylward LL, Karch NJ, Paustenbach DJ. The relative susceptibility of animals and humans to the carcinogenic hazard posed by exposure to TCDD: an analysis using standard and internal measures of dose. Chemosphere. 1997;34:1507–1522. [PubMed: 9134683] [CrossRef]
- Hébert CD, Harris MW, Elwell MR, Birnbaum LS. Relative toxicity and tumor-promoting ability of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), 2,3,4,7,8-pentachlorodibenzofuran (PCDF), and 1,2,3,4,7,8-hexachlorodibenzofuran (HCDF) in hairless mice. Toxicology and Applied Pharmacology. 1990;102:362–377. [PubMed: 2300974] [CrossRef]
- Hemming H, Bager Y, Flodström S, et al. Liver tumour promoting activity of 3,4,5,3′,4′-pentachlorobiphenyl and its interaction with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Eur J Pharmacol. 1995;292:241–249. [PubMed: 7796862]
- Hennig B, Hammock BD, Slim R, et al. PCB-induced oxidative stress in endothelial cells: modulation by nutrients. Int J Hyg Environ Health. 2002;205:95–102. [PubMed: 12018021]
- Hooiveld M, Heederik DJJ, Kogevinas M, et al. Second follow-up of a Dutch cohort occupationally exposed to phenoxy herbicides, chlorophenols, and contaminants. Am J Epidemiol. 1998;147:891–901. [PubMed: 9583720]
- Huwe JK. Dioxins in food: a modern agricultural perspective. J Agric Food Chem. 2002;50:1739–1750. [PubMed: 11902908] [CrossRef]
- IARC. Some fumigants, the herbicides 2,4-D and 2,4,5-T, chlorinated dibenzodioxins and miscellaneous industrial chemicals. IARC Monogr Eval Carcinog Risk Chem Man. 1977;15:1–354. [PubMed: 330387]
- IARC. Polychlorinated biphenyls and polybrominated biphenyls. IARC Monogr Eval Carcinog Risk Chem Hum. 1978;18:1–124. [PubMed: 215509]
- IARC. Overall evaluations of carcinogenicity: an updating of IARC Monographs volumes 1 to 42. IARC Monogr Eval Carcinog Risks Hum Suppl. 1987;7:1–440. [PubMed: 3482203]
- IARC. Polychlorinated dibenzo-para-dioxins and polychlorinated dibenzofurans. IARC Monogr Eval Carcinog Risks Hum. 1997;69:1–631. [PMC free article: PMC5366851] [PubMed: 9379504]
- Kahn PC, Gochfeld M, Nygren M, et al. Dioxins and dibenzofurans in blood and adipose tissue of Agent Orange-exposed Vietnam veterans and matched controls. JAMA. 1988;259:1661–1667. [PubMed: 3343772] [CrossRef]
- Ketchum NS, Aktar FZ (1996). The Air Force health study: an epidemiologie investigation of health effects in air force personnel following exposure to herbicides, mortality update 1996. (Interim Technical Report AL/AO-TR-1996-0068). Brooks Air Force Base, Texas: Arstrong Laboratory.
- Ketchum NS, Michalek JE, Burton JE. Serum dioxin and cancer in veterans of Operation Ranch Hand. Am J Epidemiol. 1999;149:630–639. [PubMed: 10192310]
- Knerr S, Schaefer J, Both S, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induced cytochrome P450s alter the formation of reactive oxygen species in liver cells. Mol Nutr Food Res. 2006;50:378–384. [PubMed: 16534750]
- Kociba RJ, Keyes DG, Beyer JE, et al. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicology and Applied Pharmacology. 1978;46:279–303. [PubMed: 734660] [CrossRef]
- Kogevinas M, Becher H, Benn T, et al. Cancer mortality in workers exposed to phenoxy herbicides, chlorophenols, and dioxins. An expanded and updated international cohort study. Am J Epidemiol. 1997;145:1061–1075. [PubMed: 9199536]
- Kogevinas M, Kauppinen T, Winkelmann R, et al. Soft tissue sarcoma and non-Hodgkin’s lymphoma in workers exposed to phenoxy herbicides, chlorophenols, and dioxins: two nested case-control studies. Epidemiology. 1995;6:396–402. [PubMed: 7548348]
- Kohn MC. Biochemical mechanisms and cancer risk assessment models for dioxin. Toxicology. 1995;102:133–138. [PubMed: 7482548]
- Kopec AK, Boverhof DR, Burgoon LD, et al. Comparative toxicogenomic examination of the hepatic effects of PCB126 and TCDD in immature, ovariectomized C57BL/6 mice. Toxicol Sci. 2008;102:61–75. [PubMed: 18042819]
- Kulkarni PS, Crespo JG, Afonso CAM. Dioxins sources and current remediation technologies – a review. Environ Int. 2008;34:139–153. [PubMed: 17826831] [CrossRef]
- LaKind JS, Hays SM, Aylward LL, Naiman DQ. Perspective on serum dioxin levels in the United States: an evaluation of the NHANES data. J Expo Sci Environ Epidemiol. 2009;19:435–441. [PubMed: 18854873] [CrossRef]
- Larsen JC. Risk assessments of polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, and dioxin-like polychlorinated biphenyls in food. Mol Nutr Food Res. 2006;50:885–896. [PubMed: 17009211] [CrossRef]
- Liem AK, Fürst P, Rappe C. Exposure of populations to dioxins and related compounds. Food Addit Contam. 2000;17:241–259. [PubMed: 10912239] [CrossRef]
- Lin PH, Lin CH, Huang CC, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces oxidative stress, DNA strand breaks, and poly(ADP-ribose) polymerase-1 activation in human breast carcinoma cell lines. Toxicol Lett. 2007;172:146–158. [PubMed: 17669606]
- Lu Y, Wang X, Safe S. Interaction of 2,3,7,8-tetrachlorodibenzo-p-dioxin and retinoic acid in MCF-7 human breast cancer cells. Toxicol Appl Pharmacol. 1994;127:1–8. [PubMed: 8048042]
- Lucier GW, Tritscher A, Goldsworthy T, et al. Ovarian hormones enhance 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated increases in cell proliferation and preneoplastic foci in a two-stage model for rat hepatocarcinogenesis. Cancer Res. 1991;51:1391–1397. [PubMed: 1671757]
- Luebeck EG, Buchmann A, Stinchcombe S, et al. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on initiation and promotion of GST-P-positive foci in rat liver: A quantitative analysis of experimental data using a stochastic model. Toxicol Appl Pharmacol. 2000;167:63–73. [PubMed: 10936080]
- Lynge E. Cancer in phenoxy herbicide manufacturing workers in Denmark, 1947–87–an update. Cancer Causes Control. 1993;4:261–272. [PubMed: 8318642]
- Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B. 2005;175:221–230. [PubMed: 15900503] [CrossRef]
- Manz A, Berger J, Dwyer JH, et al. Cancer mortality among workers in chemical plant contaminated with dioxin. Lancet. 1991;338:959–964. [PubMed: 1681339] [CrossRef]
- Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–1184. [PubMed: 16211578]
- Maronpot RR, Foley JF, Takahashi K, et al. Dose response for TCDD promotion of hepatocarcinogenesis in rats initiated with DEN: histologic, biochemical, and cell proliferation endpoints. Environ Health Perspect. 1993;101:634–642. [PMC free article: PMC1519881] [PubMed: 8143597]
- Martinez JM, Afshari CA, Bushel PR, et al. Differential toxicogenomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin in malignant and nonmalignant human airway epithelial cells. Toxicol Sci. 2002;69:409–423. [PubMed: 12377990]
- Mayes BA, McConnell EE, Neal BH, et al. Comparative carcinogenicity in Sprague-Dawley rats of the polychlorinated biphenyl mixtures Aroclors 1016, 1242, 1254, and 1260. Toxicol Sci. 1998;41:62–76. [PMC free article: PMC7107229] [PubMed: 9520342]
- McLean D, Eng A, Walls C, et al. Serum dioxin levels in former New Zealand sawmill workers twenty years after exposure to pentachlorophenol (PCP) ceased. Chemosphere. 2009;74:962–967. [PubMed: 19036402] [CrossRef]
- Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol Cell Biol. 1998;18:978–988. [PMC free article: PMC108810] [PubMed: 9447995]
- Michalek JE, Wolfe WH, Miner JC, et al. Indices of TCDD exposure and TCDD body burden in veterans of Operation Ranch Hand. J Expo Anal Environ Epidemiol. 1995;5:209–223. [PubMed: 7492907]
- Milbrath MO, Wenger Y, Chang CW, et al. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ Health Perspect. 2009;117:417–425. [PMC free article: PMC2661912] [PubMed: 19337517]
- Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–268. [PubMed: 12573486]
- N’Jai A, Boverhof DR, Dere E, et al. Comparative temporal toxicogenomic analysis of TCDD- and TCDF-mediated hepatic effects in immature female C57BL/6 mice. Toxicol Sci. 2008;103:285–297. [PubMed: 18343893]
- Nakatani T, Okazaki K, Ogaki S, et al. Polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, and coplanar polychlorinated biphenyls in human milk in Osaka City, Japan. Arch Environ Contam Toxicol. 2005;49:131–140. [PubMed: 15983863] [CrossRef]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. [PubMed: 15028720]
- Nebert DW, Roe AL, Dieter MZ, et al. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. [PubMed: 10605936]
- NIOSH (1990). National Occupational Exposure Survey (1981-83). Cincinnati, OH: National Institute for Occupational Safety and Health. Available at http://www
.cdc.gov/noes/noes3/empl0003 .html . - Nishizumi M, Masuda Y. Enhancing effect of 2,3,4,7,8-pentachlorodibenzofuran and 1,2,3,4,7,8-hexachlorodibenzofuran on diethylnitrosamine hepatocarcinogenesis in rats. Cancer Lett. 1986;33:333–339. [PubMed: 3802062] [CrossRef]
- NTP. Carcinogenesis Bioassay of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (CAS No. 1746–01–6) in Osborne-Mendel Rats and B6C3F1 Mice (Gavage Study). Natl Toxicol Program Tech Rep Ser. 1982;209:1–195. a. [PubMed: 12778226]
- NTP. Carcinogenesis Bioassay of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (CAS No. 1746–01–6) in Swiss-Webster Mice (Dermal Study). Natl Toxicol Program Tech Rep Ser. 1982;201:1–113. b. [PubMed: 12778178]
- NTP. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TDCC); “dioxin” Rep Carcinog. 2004;11:241–243. [PubMed: 21089963]
- NTP. NTP technical report on the toxicology and carcinogenesis studies of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (CAS No. 1746–01–6) in female Harlan Sprague-Dawley rats (Gavage Studies). Natl Toxicol Program Tech Rep Ser. 2006;521:4–232. a. [PubMed: 16835633]
- NTP. Toxicology and carcinogenesis studies of 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) (Cas No. 57117–31–4) in female Harlan Sprague-Dawley rats (gavage studies). Natl Toxicol Program Tech Rep Ser. 2006;525:1–198. b. [PubMed: 17160103]
- NTP. NTP toxicology and carcinogenesis studies of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (CAS No. 57465–28–8) in female Harlan Sprague-Dawley rats (Gavage Studies). Natl Toxicol Program Tech Rep Ser. 2006;520:4–246. c. [PubMed: 16628245]
- NTP. Toxicology and Carcinogenesis Studies of a Mixture of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) (Cas No. 1746-01-6), 2,3,4,7,8-Pentachlorodibenzofuran (PeCDF) (Cas No. 57117-31-4), and 3,3',4,4',5-Pentachlorobiphenyl (PCB 126) (Cas No. 57465-28-8) in Female Harlan Sprague-Dawley Rats (gavage studies). National Toxicology Program Technical Report Series. 2006;526:1–180. d. [PubMed: 17342195]
- NTP. 2009NTP toxicology and carcinogenesis studies of 2,3',4,4',5-pentachlorobiphenyl (PCB 118) (CAS No. 31508-00-6) in female Harlan Sprague-Dawley rats (Gavage Studies) National Toxicology Program Technical Report Series5591-174. [PubMed: 21383778]
- Olie K, Schecter A, Constable J, et al. Chlorinated dioxin and dibenzofuran levels in food and wildlife samples in the North and South of Vietnam. Chemosphere. 1989;19:493–496. [CrossRef]
- Onozuka D, Yoshimura T, Kaneko S, Furue M. Mortality after exposure to polychlorinated biphenyls and polychlorinated dibenzofurans: a 40-year follow-up study of Yusho patients. Am J Epidemiol. 2009;169:86–95. [PubMed: 18974082]
- Ott MG, Messerer P, Zober A. Assessment of past occupational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin using blood lipid analyses. Int Arch Occup Environ Health. 1993;65:1–8. [PubMed: 8354568] [CrossRef]
- Ott MG, Olson RA, Cook RR, Bond GG. Cohort mortality study of chemical workers with potential exposure to the higher chlorinated dioxins. J Occup Med. 1987;29:422–429. [PubMed: 2439670]
- Ott MG, Zober A. Cause specific mortality and cancer incidence among employees exposed to TCDD after a 1953 reactor accident. Occup Environ Med. 1996;53:606–612. [PMC free article: PMC1128557] [PubMed: 8882118] [CrossRef]
- Parzefall W. Risk assessment of dioxin contamination in human food. Food Chem Toxicol. 2002;40:1185–1189. [PubMed: 12067582] [CrossRef]
- Pastorelli R, Carpi D, Campagna R, et al. Differential expression profiling of the hepatic proteome in a rat model of dioxin resistance: correlation with genomic and transcriptomic analyses. Molecular & Cellular Proteomics. 2006;5 (5):882–894. [PubMed: 16497791]
- Peng TL, Chen J, Mao W, et al. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009;10:27. [PMC free article: PMC2680824] [PubMed: 19371443]
- Pesatori AC, Consonni D, Rubagotti M, et al. Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ Health. 2009;8:39. [PMC free article: PMC2754980] [PubMed: 19754930] [CrossRef]
- Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem. 1976;251:4936–4946. [PubMed: 956169]
- Portier C, Kohn M. A biologically-based model for the carcinogenic effects of TCDD in female Sprague-Dawley rats. Organohalogen Compounds. 1996;29::222–227.
- Portier CJ, Sherman CD, Kohn M, et al. Modeling the number and size of hepatic focal lesions following exposure to TCDD. Toxicol Appl Pharmacol. 1996;138:20–30. [PubMed: 8658509]
- Rao MS, Subbarao V, Prasad JD, Scarpelli DG. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the Syrian golden hamster. Carcinogenesis. 1988;9:1677–1679. [PubMed: 3409472] [CrossRef]
- Ray S, Swanson HI. Activation of the aryl hydrocarbon receptor by TCDD inhibits senescence: a tumor promoting event? Biochem Pharmacol. 2009;77:681–688. [PMC free article: PMC2662439] [PubMed: 19100242]
- Ryan JJ, Schecter A. Exposure of Russian phenoxy herbicide producers to dioxins. J Occup Environ Med. 2000;42:861–870. [PubMed: 10998761] [CrossRef]
- Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett. 2001;120:1–7. [PubMed: 11323156]
- Safe S, Wormke M. Inhibitory aryl hydrocarbon receptor-estrogen receptor alpha cross-talk and mechanisms of action. Chem Res Toxicol. 2003;16:807–816. [PubMed: 12870882]
- Saracci R, Kogevinas M, Bertazzi PA, et al. Cancer mortality in workers exposed to chlorophenoxy herbicides and chlorophenols. Lancet. 1991;338:1027–1032. [PubMed: 1681353] [CrossRef]
- Sasamoto T, Ushio F, Kikutani N, et al. Estimation of 1999–2004 dietary daily intake of PCDDs, PCDFs and dioxin-like PCBs by a total diet study in metropolitan Tokyo, Japan. Chemosphere. 2006;64:634–641. [PubMed: 16376969] [CrossRef]
- Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: an overview. Environ Res. 2006;101:419–428. [PubMed: 16445906] [CrossRef]
- Schecter A, Dai LC, Päpke O, et al. Recent dioxin contamination from Agent Orange in residents of a southern Vietnam city. J Occup Environ Med. 2001;43:435–443. [PubMed: 11382178] [CrossRef]
- Schecter A, McGee H, Stanley J, Boggess K. Dioxin, dibenzofuran, and PCB including coplanar PCB levels in the blood of Vietnam veterans in the Michigan Agent Orange Study. Chemosphere. 1992;25:205–208. [CrossRef]
- Schecter A, Pavuk M, Constable JD, et al. A follow-up: high level of dioxin contamination in Vietnamese from agent orange, three decades after the end of spraying. J Occup Environ Med. 2002;44:218–220. [PubMed: 11911020] [CrossRef]
- Schecter A, Quynh HT, Pavuk M, et al. Food as a source of dioxin exposure in the residents of Bien Hoa City, Vietnam. J Occup Environ Med. 2003;45:781–788. [PubMed: 12915779] [CrossRef]
- Schecter A, Ryan JJ, Constable JD, et al. Partitioning of 2,3,7,8-chlorinated dibenzo-p-dioxins and dibenzofurans between adipose tissue and plasma lipid of 20 Massachusetts Vietnam veterans. Chemosphere. 1990;20:951–958. [CrossRef]
- Schecter A, Startin J, Wright C, et al. Congener-specific levels of dioxins and dibenzofurans in U.S. food and estimated daily dioxin toxic equivalent intake. Environ Health Perspect. 1994;102:962–966. [PMC free article: PMC1567464] [PubMed: 9738211] [CrossRef]
- Schlezinger JJ, Struntz WD, Goldstone JV, Stegeman JJ. Uncoupling of cytochrome P450 1A and stimulation of reactive oxygen species production by co-planar polychlorinated biphenyl congeners. Aquat Toxicol. 2006;77:422–432. [PubMed: 16500718]
- Schwanekamp JA, Sartor MA, Karyala S, et al. Genome-wide analyses show that nuclear and cytoplasmic RNA levels are differentially affected by dioxin. Biochim Biophys Acta. 2006;1759:388–402. [PubMed: 16962184]
- Schwarz M, Appel KE. Carcinogenic risks of dioxin: mechanistic considerations. Regul Toxicol Pharmacol. 2005;43:19–34. [PubMed: 16054739]
- Scott LLF, Unice KM, Scott P, et al. Addendum to: Evaluation of PCDD/F and dioxin-like PCB serum concentration data from the 2001–2002 National Health and Nutrition Examination Survey of the United States population. J Expo Sci Environ Epidemiol. 2008;18:524–532. [PubMed: 18368012]
- Shertzer HG, Nebert DW, Puga A, et al. Dioxin causes a sustained oxidative stress response in the mouse. Biochem Biophys Res Commun. 1998;253:44–48. [PubMed: 9875217]
- Silkworth JB, Koganti A, Illouz K, et al. Comparison of TCDD and PCB CYP1A induction sensitivities in fresh hepatocytes from human donors, sprague-dawley rats, and rhesus monkeys and HepG2 cells. Toxicol Sci. 2005;87:508–519. [PubMed: 16049271]
- Smart J, Daly AK. Variation in induced CYP1A1 levels: relationship to CYP1A1, Ah receptor and GSTM1 polymorphisms. Pharmacogenetics. 2000;10:11–24. [PubMed: 10739168]
- Smith AH, Patterson DG Jr, Warner ML, et al. Serum 2,3,7,8-tetrachlorodibenzo-p-dioxin levels of New Zealand pesticide applicators and their implication for cancer hypotheses. J Natl Cancer Inst. 1992;84:104–108. [PubMed: 1735875] [CrossRef]
- Steenland K, Bertazzi P, Baccarelli A, Kogevinas M. Dioxin revisited: developments since the 1997 IARC classification of dioxin as a human carcinogen. Environ Health Perspect. 2004;112:1265–1268. [PMC free article: PMC1247514] [PubMed: 15345337]
- Steenland K, Deddens J, Piacitelli L. Risk assessment for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) based on an epidemiologic study. Am J Epidemiol. 2001;154:451–458. [PubMed: 11532787] [CrossRef]
- Steenland K, Piacitelli L, Deddens J, et al. Cancer, heart disease, and diabetes in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Natl Cancer Inst. 1999;91:779–786. [PubMed: 10328108] [CrossRef]
- Stohs SJ, Shara MA, Alsharif NZ, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced oxidative stress in female rats. Toxicol Appl Pharmacol. 1990;106:126–135. [PubMed: 2251677]
- Thiess AM, Frentzel-Beyme R, Link R. Mortality study of persons exposed to dioxin in a trichlorophenol-process accident that occurred in the BASF AG on November 17, 1953. Am J Ind Med. 1982;3:179–189. [PubMed: 6215858] [CrossRef]
- Thornton AS, Oda Y, Stuart GR, et al. Mutagenicity of TCDD in Big Blue transgenic rats. Mutat Res. 2001;478:45–50. [PubMed: 11406168]
- Todaka T, Hirakawa H, Kajiwara J, et al. Concentrations of polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, and dioxin-like polychlorinated biphenyls in blood and breast milk collected from 60 mothers in Sapporo City, Japan. Chemosphere. 2008;72:1152–1158. [PubMed: 18474391] [CrossRef]
- Tóth K, Somfai-Relle S, Sugár J, Bence J. Carcinogenicity testing of herbicide 2,4,5-trichlorophenoxyethanol containing dioxin and of pure dioxin in Swiss mice. Nature. 1979;278:548–549. [PubMed: 431718] [CrossRef]
- Toyoshiba H, Yamanaka T, Sone H, et al. Gene interaction network suggests dioxin induces a significant linkage between aryl hydrocarbon receptor and retinoic acid receptor beta. Environ Health Perspect. 2004;112:1217–1224. [PMC free article: PMC1277115] [PubMed: 15345368]
- Tritscher AM, Clark GC, Sewall C, et al. Persistence of TCDD-induced hepatic cell proliferation and growth of enzyme altered foci after chronic exposure followed by cessation of treatment in DEN initiated female rats. Carcinogenesis. 1995;16:2807–2811. [PubMed: 7586202]
- Tritscher AM, Mahler J, Portier CJ, et al. Induction of lung lesions in female rats following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Pathol. 2000;28:761–769. [PubMed: 11127289]
- Tritscher AM, Seacat AM, Yager JD, et al. Increased oxidative DNA damage in livers of 2,3,7,8-tetrachlorodibenzo-p-dioxin treated intact but not ovariectomized rats. Cancer Lett. 1996;98:219–225. [PubMed: 8556712]
- Tsai PC, Ko YC, Huang W, et al. Increased liver and lupus mortalities in 24-year follow-up of the Taiwanese people highly exposed to polychlorinated biphenyls and dibenzofurans. Sci Total Environ. 2007;374:216–222. [PubMed: 17257654]
- Tsutsumi T, Yanagi T, Nakamura M, et al. Update of daily intake of PCDDs, PCDFs, and dioxin-like PCBs from food in Japan. Chemosphere. 2001;45:1129–1137. [PubMed: 11695626] [CrossRef]
- Tuomisto JT, Viluksela M, Pohjanvirta R, Tuomisto J. The AH receptor and a novel gene determine acute toxic responses to TCDD: segregation of the resistant alleles to different rat lines. Toxicol Appl Pharmacol. 1999;155:71–81. [PubMed: 10036220]
- USEPA (2000a). Exposure and Human Health Reassessment of 2,3,7,8-Tetrachlorodiobenzo-p-dioxin (TCDD) and Related Compounds (September 2000 Draft). Part I: Estimating Exposure to dioxin-like compounds. Volume 2: Sources of dioxin-like compounds in the United States. EPA/600/P-00/001 Bb. U.S. Environmental Protection Agency, Washington, DC: National Center for Environmental Assessment, Office of Research and Development.
- USEPA (2000b). Exposure and Human Health Reassessment of 2,3,7,8-Tetrachlorodiobenzo-p-dioxin (TCDD) and Related Compounds (September 2000 Draft). Part I: Estimating Exposure to dioxin-like compounds. Volume 3: Properties, environmental levels and background exposures. EPA/600/P-00/001 Bc. U.S. Environmental Protection Agency, Washington, DC: National Center for Environmental Assessment, Office of Research and Development.
- USEPA (2004). Exposure and human health reassessment of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. National Academy of Sciences Review Draft. Washington, DC: National Center for Environmental Assessment, Office of Research and Development. Available at http://www
.epa.gov/ncea /pdfs/dioxin/nas-review. - USEPA (2006). An Inventory of Sources and Environmental Releases of Dioxin-Like Compounds in the United States for the Years 1987, 1995, and 2000. Washington, DC: National Center for Environmental Assessment, Office of Research and Development. Available at http://www
.epa.gov/ncea/pdfs/dioxin/ - Van den Berg M, Birnbaum LS, Denison M, et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol Sci. 2006;93:223–241. [PMC free article: PMC2290740] [PubMed: 16829543]
- Van Miller JP, Lalich JJ, Allen JR. lncreased incidence of neoplasms in rats exposed to low levels of 2,3,7, 8-tetrachlorodibenzo-p-dioxin. Chemosphere. 1977;9:537–544. [CrossRef]
- Vezina CM, Walker NJ, Olson JR. Subchronic exposure to TCDD, PeCDF, PCB126, and PCB153: effect on hepatic gene expression. Environ Health Perspect. 2004;112:1636–1644. [PMC free article: PMC1247661] [PubMed: 15598615]
- Viluksela M, Bager Y, Tuomisto JT, et al. Liver tumor-promoting activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in TCDD-sensitive and TCDD-resistant rat strains. Cancer Res. 2000;60:6911–6920. [PubMed: 11156390]
- Vondrácek J, Machala M, Bryja V, et al. Aryl hydrocarbon receptor-activating polychlorinated biphenyls and their hydroxylated metabolites induce cell proliferation in contact-inhibited rat liver epithelial cells. Toxicol Sci. 2005;83:53–63. [PubMed: 15483185]
- Waern F, Flodström S, Busk L, et al. Relative liver tumour promoting activity and toxicity of some polychlorinated dibenzo-p-dioxin- and dibenzofuran-congeners in female Sprague-Dawley rats. Pharmacol Toxicol. 1991;69:450–458. [PubMed: 1766921] [CrossRef]
- Walker NJ. Unraveling the complexities of the mechanism of action of dioxins. Toxicol Sci. 2007;95:297–299. [PubMed: 17297723]
- Walker NJ, Crockett PW, Nyska A, et al. Dose-additive carcinogenicity of a defined mixture of “dioxin-like compounds”. Environ Health Perspect. 2005;113:43–48. [PMC free article: PMC1253708] [PubMed: 15626646]
- Walker NJ, Yoshizawa K, Miller RA, et al. Pulmonary lesions in female Harlan Sprague-Dawley rats following two-year oral treatment with dioxin-like compounds. Toxicol Pathol. 2007;35:880–889. [PMC free article: PMC2633090] [PubMed: 18098034]
- Wyde ME, Braen AP, Hejtmancik M, et al. Oral and dermal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces cutaneous papillomas and squamous cell carcinomas in female hemizygous Tg.AC transgenic mice. Toxicological Sciences. 2004;82:34–45. [PubMed: 15282402] [CrossRef]
- Wyde ME, Wong VA, Kim AH, et al. Induction of hepatic 8-oxo-deoxyguanosine adducts by 2,3,7,8-tetrachlorodibenzo-p-dioxin in Sprague-Dawley rats is female-specific and estrogen-dependent. Chem Res Toxicol. 2001;14:849–855. [PubMed: 11453731]
- Yoshida R, Ogawa Y. Oxidative stress induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin: an application of oxidative stress markers to cancer risk assessment of dioxins. Ind Health. 2000;38:5–14. [PubMed: 10680305]
- Yoshizawa K, Walker NJ, Jokinen MP, et al. Gingival carcinogenicity in female Harlan Sprague-Dawley rats following two-year oral treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and dioxin-like compounds. Toxicological Sciences. 2005;83:64–77. [PubMed: 15509667] [CrossRef]
- Zack JA, Gaffey WR (1983). A mortality study of workers employed at the Monsanto Company plant in Nitro, West Virginia. In: Human and Environmental Risks of Chlorinated Dioxins and Related Compounds. Tucker R, Young A, Grey A, editors. New York: Plenum Press, pp. 575–591.
- Zack JA, Suskind RR. The mortality experience of workers exposed to tetrachlorodibenzodioxin in a trichlorophenol process accident. J Occup Med. 1980;22:11–14. [PubMed: 6444441] [CrossRef]
- Zook DR, Rappe C (1994) Environmental sources, distribution, and fate of polychlorinated dibenzodioxins, dibenzofurans, and related organochlorines. In: Schecter, A., ed., Dioxins and Health. New York, Plenum Press, pp. 80-113.
- Zubero MB, Ibarluzea JM, Aurrekoetxea JJ, et al. Serum levels of polychlorinated dibenzodioxins and dibenzofurans and PCBs in the general population living near an urban waste treatment plant in Biscay, Basque Country. Chemosphere. 2009;76:784–791. [PubMed: 19482333]
- 2,3,7,8-TETRACHLORODIBENZO-para-DIOXIN, 2,3,4,7,8-PENTACHLORODIBENZOFURAN, AND 3...2,3,7,8-TETRACHLORODIBENZO-para-DIOXIN, 2,3,4,7,8-PENTACHLORODIBENZOFURAN, AND 3,3′,4,4′,5-PENTACHLOROBIPHENYL - Chemical Agents and Related Occupations
Your browsing activity is empty.
Activity recording is turned off.
See more...