

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Center for Biotechnology Information (US). Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1998-.

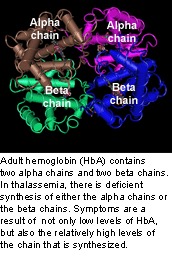
Thalassemia is an inherited disease of faulty synthesis of hemoglobin. The name is derived from the Greek word "thalassa" meaning "the sea" because the condition was first described in populations living near the Mediterranean Sea; however, the disease is also prevalent in Africa, the Middle East, and Asia.
Thalassemia consists of a group of disorders that may range from a barely detectable abnormality of blood, to severe or fatal anemia. Adult hemoglobin is composed of two alpha (α) and two beta (β) polypeptide chains. There are two copies of the hemoglobin alpha gene (HBA1 and HBA2), which each encode an α-chain, and both genes are located on chromosome 16. The hemoglobin beta gene (HBB) encodes the β-chain and is located on chromosome 11.
In α-thalassemia, there is deficient synthesis of α-chains. The resulting excess of β-chains bind oxygen poorly, leading to a low concentration of oxygen in tissues (hypoxemia). Similarly, in β-thalassemia there is a lack of β-chains. However, the excess α-chains can form insoluble aggregates inside red blood cells. These aggregates cause the death of red blood cells and their precursors, causing a very severe anemia. The spleen becomes enlarged as it removes damaged red blood cells from the circulation.
Deletions of HBA1 and/or HBA2 tend to underlie most cases of α-thalassemia. The severity of symptoms depends on how many of these genes are lost. Loss of one or two genes is usually asymptomatic, whereas deletion of all four genes is fatal to the unborn child.
In contrast, over 100 types of mutations affect HBB, and deletion mutations are rare. Splice mutations and mutations that occur in the HBB gene promoter region tend to cause a reduction, rather than a complete absence, of β-globin chains and so result in milder disease. Nonsense mutations and frameshift mutations tend to not produce any β-globin chains leading to severe disease.
Currently, severe thalassemia is treated by blood transfusions, and a minority of patients are cured by bone marrow transplantation. Mouse models are proving to be useful in assessing the potential of gene therapy.
Related diseases
- Genome view see gene locations
- Entrez Gene collection of gene-related information
- BLink α-chain
- BLink β-chain related sequences in different organisms
- Research articles online full text
- Books online books section
- OMIM catalog of human genes and disorders
- Cooley's Anemia FoundationFurther information for patients and families
- Thalassemia.ComFurther information for patients and health care workers
- OMIMRelated OMIM records
- Thalassemia - Genes and DiseaseThalassemia - Genes and Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...