Clinical Description
Mucolipidosis III alpha/beta (ML III alpha/beta; pseudo-Hurler polydystrophy) is a slowly progressive inborn error of metabolism with clinical onset at approximately age three years and fatal outcome in early to middle adulthood [Leroy 2007, Cathey et al 2010]. Comprehensive data on life expectancy are still lacking.
Growth. Weight and length at birth are within normal limits. Gradual slowing of growth rate begins in late infancy to early childhood. Concerns about small stature rarely arise before age three years, when worsening shoulder, hip, and knee contractures adversely affect stature. ML III alpha/beta does not cause frank dwarfism as does ML II (see ); however, stature from early childhood is often below the third centile on standard growth curves (see , ). Final stature is well below expected for an individual’s average family stature.
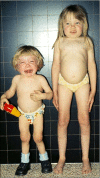
Girl (age 9.5 yrs) on the right has mucolipidosis type III alpha/beta. Boy (age 3 yrs) on the left has mucolipidosis II. Hands in the two children are significantly different: short, broad with claw-like in ML II and rather long in ML III alpha/beta. (more...)
A. Deficient linear growth in ML III alpha/beta is illustrated by the difference in stature in dizygotic twin girls. The affected twin is shown on the right. B. Growth of the affected twin (solid circles) and her healthy twin (open circles) plotted on (more...)
Craniofacial. True macrocephaly does not occur. Dysmorphic facial features are absent or minimal in younger children. Coarsening of facial features is gradual and more apparent in profile, including full cheeks, depressed nasal bridge, and prominent mouth. Gingival hypertrophy is mild and does not usually interfere with tooth eruption.
Ophthalmologic. Epicanthal folds persist longer than normal. Proptosis, often observed in ML II, is rare. The corneas are clear by routine clinical inspection, but opacities may be appreciated by slit-lamp examination.
Audiologic. Episodes of otitis media occur in individuals with ML III alpha/beta more frequently than in the general population. Conductive hearing loss, documented in some affected individuals, has not been studied systematically. Sensorineural hearing loss is not a typical feature of ML III.
Respiratory. Mild hoarseness of the voice is an inconsistent finding. Upper-respiratory infections are more frequent than expected in some (but not all) children. From late childhood bronchitis and bronchopneumonia are the most consistent clinical complications.
Adults exhibit restrictive lung disease caused by stiffening of the thoracic cage, slowly progressive sclerosis of bronchi, and hardening and thickening of the interstitial tissue (extracellular matrix) in lung parenchyma.
Cardiovascular. Individuals with ML III alpha/beta are at risk for cardiac involvement. Gradual thickening and subsequent insufficiency of the mitral valve and the aortic valve are common from late childhood onward [Steet et al 2005].
Left and/or right ventricular hypertrophy is often documented on echocardiography in older individuals. Pulmonary hypertension may occur in some older individuals, but at present is still insufficiently documented.
Rapid progression of cardiac disease is rarely observed in ML III alpha/beta.
Pneumonia may compound mild cardiac insufficiency. Death in early adulthood is often from cardiopulmonary causes, even without complicating factors such as pneumonia.
Gastrointestinal. Prominence of the abdomen especially upon standing upright is caused in part by lumbar hyperlordosis, compensation for hip and knee flexion contractures, and hypotonia of the abdominal wall musculature. Diastasis of the medial recti and small umbilical hernias may also be present. In general, individuals with ML III alpha/beta do not present with organomegaly.
Skeletal/ soft connective tissue. Stiffness of all large and small joints is a cardinal feature. Limited range of motion in the shoulders is frequently the initial evidence of ML III alpha/beta and is mainly of soft tissue origin.
Limited range of motion in the hips and knees explains the slow gait and inability of children to run effectively. Flexion contractures in the hips and knees cause the squatting standing posture, most apparent in lateral view (see ).
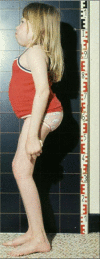
Same patient with ML III alpha/beta as in Figure 2 at age 12 years. Profile view shows posture adversely affected by flexion contractures and stiffness in the hips and knees with compensatory dorsal hyperlordosis and sacral hyperkyphosis. Hands and fingers (more...)
Secondary but severe arthritic changes in the hips that can lead to destruction of the proximal femoral epiphyses make walking increasingly difficult and painful. Significant hardening of the surrounding soft tissues contributes to hip dysfunction. Many affected individuals become wheelchair bound before or during early adulthood.
Range of motion in the wrists and ankles is less adversely affected, than in the other large joints. Dupuytren-type palmar contractures may appear from late childhood onward and exacerbate the moderate to severe claw-like flexion deformity of the fingers associated with recurrent swelling and progressive stiffness. Neuropathic carpal tunnel signs can become severe in some individuals.
In ML III alpha/beta the hands and fingers are usually of near-normal length in contrast to the severely affected hands in ML II.
Before the appropriate diagnosis is made, many individuals with ML III alpha/beta have been evaluated for a rheumatologic disorder.
Osteoporosis affects the entire skeleton. Bone pain becomes the most distressing symptom in Ml III alpha/beta, even in individuals with limited ambulation. Osteolytic bone lesions also are associated with significant bone pain in those who are non-ambulatory.
Neuromotor development and intellect are the most variable features in ML III alpha/beta, ranging from normal to mild or moderate developmental delay in reaching motor milestones. Onset and development of receptive and expressive language skills occur at the expected age. Stuttering has not been observed in individuals with ML III alpha/beta. Although psychometric tests often reveal an IQ within normal limits, approximately half of the affected children require school assistance, often because of their physical limitations.
Other. The neck is short. Thickening of the skin is inconsistent and mild.
Previously used diagnostic testing. Phase-contrast or electron microscopic (EM) demonstration of large amounts of dense cytoplasmic inclusions (I-cells) in cultured fibroblasts was previously used to help confirm the diagnosis of ML II and ML III alpha/beta (see ).
Living culture of skin fibroblasts derived from a person with ML III alpha/beta viewed by the contrast light microscope. The cytoplasm is filled with dense granular inclusions that consistently spare a juxtanuclear zone that represents the endoplasmic (more...)
Note: On electron microscopy (EM) the mesenchymal cells in any tissue reveal large numbers of cytoplasmic vacuoles comprising swollen lysosomes bound by a unit membrane. The contents are pleomorphic, but not dense. This phenomenon is specific to ML II and ML III alpha/beta and is not observed in any lysosomal storage disorder.
The activity of lysosomal enzymes is severely reduced in I-cells, but significantly increased in the corresponding culture media.
The cytologic and enzymatic findings in cell culture cannot distinguish between ML II (I-cell disease) and ML III alpha/beta (see ML II).
Nomenclature
Pseudo-Hurler-polydystrophy (PHP) was the term used in 1966 by Maroteaux and Lamy when they first clinically and radiologically delineated the multisystem disorder that had only progressive stiffening of the large and small joints in common with Hurler disease or mucopolysaccharidosis I (MPS I)
Mucolipidosis (ML). The term PHP has been largely replaced by the term mucolipidosis III, introduced in 1970 by Spranger and Wiedemann, who provided the first clinical classification of the group of metabolic disorders clinically intermediate between the lipidoses and the MPSs (storage disorders of glycosaminoglycans). Their hypothesis that some of these disorders could be pathogenetically and genetically related was confirmed in 1973 when the alignment of ML III (PHP) and ML II (I-cell disease) was shown by the discovery of the "in vitro" cytologic and biochemical I-cell phenomenon in PHP fibroblasts.
Although mucolipidosis is a clinically useful name, biochemists consider it a misnomer because "mucolipids" do not exist in nature. The term mucolipidosis has been used in four different inborn errors of metabolism; only ML II and ML III alpha/beta are GNPTAB related. Mucolipidosis I (also called sialidosis type II) and mucolipidosis IV, are genetically distinct disorders.
Oligosaccharidoses (OSs). During the 1970s, excessive urinary excretion of OSs was documented in most of the mucolipidoses; therefore, the term "oligosaccharidoses" and later the term "glycoproteinoses" have been substituted for the term mucolipidoses.
Mucolipidosis II, mucolipidosis III alpha/beta, and mucolipidosis III gamma. Because even the trivial name of the causal enzyme defect UDP-N-acetylglucosamine: lysosomal hydrolase N-acetylglucosamine 1-phosphotransferase is long, the current naming of ML II and ML III alpha/beta as "UPDGlcNAc 1-P-transferase deficiency disorders" is cumbersome; it is, however, strictly the most correct one [Leroy 2007].
The enzyme GNPT is the product of two genes, one encoding the alpha and beta subunits, and the other encoding the gamma subunit [Bao et al 1996]: