

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Osteoporosis is a multifactorial disorder associated with low bone mass and enhanced skeletal fragility. Although most prevalent in older females, some men are also at high risk. Risk factors in men and women include smoking, family history of fracture, age greater than 65 years, and low but also high BMI particularly in men. Secondary causes of osteoporosis include chronic treatment with glucocorticoids, gastrointestinal disorders, diabetes mellitus (T1D, T2D), rheumatoid arthritis, liver disease, gluten enteropathy, multiple myeloma and other hematologic disorders. However, primary osteoporosis is most often related to either postmenopausal estrogen loss or age-related deterioration of skeletal microarchitecture; both are due to uncoupling in the bone remodeling unit. Reduced bone formation with age is almost certainly a function of impaired stem cell differentiation into the osteoblast lineage with a resultant increase in marrow adipogenesis. Increased bone resorption also characterizes most forms of osteoporosis but the etiology is multifactorial. Changes in local and systemic growth factors are often responsible for uncoupling between resorption and formation. However, alterations in peak bone acquisition contribute years later to low bone mass and enhanced skeletal fragility. Fracture risk assessment tools (e.g. FRAX) in handheld apps and computers which combine bone density score and risk factors, have provided rapid assessments of future osteoporotic fractures and can be performed at the bedside. Newer methods of measuring bone quality have led to insights into micro-architectural deterioration that contributes to skeletal fragility. Notwithstanding, low areal bone mineral density by DEXA remains the strongest predictor of subsequent fracture beyond age, and this is potentially measurable in everyone after age 65. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Osteoporosis is a disorder characterized by reduced bone mass, impaired bone quality, and a propensity to fracture. For decades, this disease was considered a syndrome characterized by back pain, vertebral fractures, and osteopenia on plain films. Identifying secondary causes of low bone mass was the principle objective of most clinicians. However, osteoporosis is now classified as a primary disorder of the skeleton related to profound metabolic changes not only in bone but also related to changes in whole body homeostasis. Significant progress has been made in both defining this disorder and in understanding its complex pathogenesis. In addition, a consensus has emerged concerning the strength of the association between low bone mineral density and fracture risk, and the importance of qualitative aspects of the skeleton, as additional risk determinants.
Dual energy x-ray absorptiometry (subsequently referred to as DEXA) revolutionized our ability to predict fractures in large numbers of subjects by measuring areal bone mineral density (subsequently referred to as BMD). Virtually all population studies have confirmed that for every one standard deviation below young normal mean bone mineral density (at virtually any skeletal site) there is a nearly two-fold greater risk of a subsequent hip fracture (1). Some clinicians, as well as the WHO and the National Osteoporosis Foundation define osteoporosis purely on a bone mineral density (BMD) T-score more than 2.5 standard deviations below young normal reference ranges for the spine, hip or radius (2). Although this "bar" has been used to establish prevalence estimates and to define high risk individuals who should be considered for treatment, it is also evident that even this definition demands a better understanding of the pathophysiologic processes that result in low bone mass, and a more thorough review of 'bone quality'. Indeed, despite the strength of the association between BMD and fracture risk, qualitative measurements of the skeleton, such as bone turnover, mineralization, and trabecular connectivity also contribute to risk. In this chapter, the mechanisms responsible for altering bone microarchitecture and strength in such a way as to enhance the likelihood of fragility fractures will be reviewed. Irrespective of the epidemiology and pathogenesis of osteoporosis, the stark fact remains that this disease has significant morbidity, mortality, and economic costs. Just as importantly, understanding how this disorder develops and progresses, has important socio- economic as well as medical consequences.
EPIDEMIOLOGY OF OSTEOPOROSIS
Estimating exactly how many women have osteoporosis depends on the working definition of this disease and the appropriate diagnostic criteria (3). Prior to the widespread application of DEXA, osteoporosis was rarely diagnosed and then only in women with symptomatic vertebral fractures or osteopenia noted by x-ray for other reasons. Indeed, for too long, hip fractures, the end point of this metabolic syndrome, were either written off as a consequence of aging, or ignored in respect to treatment. BMD measurements by DEXA and CT changed all that, especially when it became clear that a single BMD measurement at any site was a very strong predictor of future spine and hip fractures (1,2,4). As such, the definition of osteoporosis began to evolve, and estimates of how many people were affected also changed. When the World Health Organization (WHO) set a cut off point of 2.5 standard deviations below a young normal mean value for BMD in the spine or hip of postmenopausal women, as an indicator of osteoporotic risk, estimates of disease prevalence increased (5). These were confirmed by publication of larger epidemiologic studies in women such as The Study of Osteoporotic Fractures (SOF) and MrOs, a large international cohort of men which provided better estimates of disease prevalence, onset and clinical course (4,6).
Currently most estimates suggest that there are approximately 0.3 million hip fractures per annum in the U.S. and 1.7 million hip fractures in Europe (7,8). With the introduction of readily available treatments, and clear prevention messaging annual hip fracture rates in the early 2000s started to decline. However, by 2015 those rates had flattened out and were trending upwards, following widespread reporting of atypical femoral fractures in patients treated with bisphosphonates and denosumab. Virtually all hip fractures can be attributed to osteoporosis, whether primary or secondary. Moreover, in most if not all cases, falls are a primary event leading to the fracture. The female to male ratio of hip fractures is approximately 2:1.0 (6,9). Not surprisingly, the occurrence of these fractures increases exponentially with age. In contrast, the incidence of wrist fractures in the UK and the US ranges from about 400- 800 per 100,000 women but is relatively stable over several decades of older life (9). Women are far more likely to suffer a Colle's fracture than a man (i.e. a ratio upwards of 10:1 by age 75) (9). Compression fractures of the vertebrae are much more difficult to estimate because often these can be asymptomatic. Best estimates are that more than a million American postmenopausal women will suffer a spine fracture in the course of a single year (9-11). The female to male ratio of occurrence is approximately 2:1. Moreover, both symptomatic, and radiographic (morphometric) fractures are associated with significant morbidity and disability (10). Finally, estimates about disease prevalence in women and men without fractures, but with low BMD (-2.5 or lower) vary greatly, but place the overall number at close to 25 million Americans and many more world-wide (9,12).
Bone loss as a result of aging/and or estrogen deficiency is the predominant pathophysiologic disorder of primary osteoporosis. However, the frequent use of glucocorticoids in both men and women, contribute dramatically to the total number of individuals with very low BMD and/or osteoporotic fractures (11). It is estimated that more than 5 million American men are afflicted with osteoporosis, based on either the presence of osteoporotic fractures (i.e. vertebral compressions, wrist fractures, hip fractures, or humerus/tibial fractures) or low BMD (11). However, the number of cohort studies in men with this disease is somewhat limited, and a more complete epidemiologic picture of male osteoporosis is becoming clearer with the MrOS study. Importantly, and somewhat surprisingly, in this cohort of more than 5000 men over the age of 65, obesity was associated with a significantly increased risk of fracture (11-13).
More frightening than estimates of the extent of low bone mass is the concern about the lower frequency of diagnosing osteoporosis and the poor adherence to therapy. It is now estimated that more than 70% of individuals who are at risk for osteoporosis, and who are receiving therapy, will not continue beyond the first year (14). Moreover, prescribing rates for bisphosphonates have fallen significantly due to the perceived risk of atypical femoral fractures (15,16). Whereas, osteoporosis was once considered a disorder of old Northern European women, it is now clear that this disease can occur in postmenopausal African Americans, that it is much more common in men than previously appreciated, and that the use of glucocorticoids and/or immunosuppressive therapies for transplant patients, markedly enhances that risk. Interestingly, and somewhat alarmingly although the prevalence of hip fractures has increased slightly, (possibly due to poor compliance from the perceived adverse events with anti-osteoporosis therapy) the diagnosis of osteoporosis by primary physicians has dropped.
FACTORS THAT AFFECT BONE QUANTITY AND QUALITY
Several risk factors predispose individuals to osteoporotic fractures. For a hip fracture, these include age greater than 65 years, a previous spine or hip fracture, maternal history of a hip fracture, poor neuromuscular function, weight loss after the age of 50, and low body mass index (1,18) (See Table 1). Falls are a major cause of fractures, and in all clinical situations, some degree of trauma can be linked to the injury (19). But many osteoporotic patients suffer fractures with minimal trauma, and this feature is pathognomonic of the skeletal fragility which accompanies low bone mass. It is for this reason that the most significant risk factor for fractures of the spine, hip or wrist remains low BMD (20). This association has been confirmed by use of the FRAX score (www.shef.ac.uk/frax) that includes areal BMD plus clinical risk factors including family history, BMI, smoking and use of glucocorticoids (21). The continuous but inverse relationship between BMD and fracture is consistent at all points below the mean suggesting there is no threshold effect (18,19). And, it is applicable at virtually every skeletal site by multiple types of measurements from the spine to finger to the calcaneus. Moreover, the advent of newer technology to measure bone mass has allowed widespread screening for risk as well as defining risk reduction with therapy. Notwithstanding the strength of the inverse relationship of BMD to fracture risk, it is important to note that the presence of a previous fracture is also an extremely important, BMD-independent risk for subsequent fracture. This is relevant clinically since the recognition of fracture at any BMD defines a skeleton that has poor bone quality, and hence is likely to fracture again.
Table 1.
Phenotypic Characteristics that Adversely Affect Bone Strength
| Characteristic | Recognized Risk Factor for Fracture | Clinically Measurable |
|---|---|---|
| Bone Mineral Density (areal) | Yes | Yes (DXA) |
| Bone Mineral Density (volumetric) | Yes | Yes (QCT) |
| Microcracks | No | No (Histology) |
| Trabecular Connectivity | Yes | TBS (DXA) |
| Periosteal Circumference | No | Yes (pQCT/DXA) |
| Cortical Thickness | +/- | Yes (pQCT/DXA) |
| Bone Turnover | Yes | Yes (markers) |
| Previous Osteoporotic Fracture | Yes | Yes (radiograms or IVA) |
| Mineralization and cortical porosity | ? | No/yes (back scatter EM) and sometimes with high resolution microCT |
IVA- instant vertebral assessment- DXA scan for morphometric changes in the thoracic and lumbar spine.
Bone mass measurement defines mineral content per area of bone. In the laboratory, bone density by DEXA is a very strong predictor of bone strength and accounts for about 80% of the variability in the breaking strength of a single femur. Thus, a very low BMD can be linked to increased skeletal fragility with a great degree of confidence. Indeed, the FRAX tool has been independently validated as the most accurate tool to measure fracture risk and includes bone mineral density as a major component (21). But there are other determinants of bone strength, often referred to as qualitative measures, including the rate of bone turnover, the extent of trabecular connectivity, cortical and periosteal bone size, and skeletal morphometry (See Figure 1 and Table 1). Indeed, much progress has been made in quantifying several aspects of bone 'quality' utilizing tools such as single energy QCT of the spine/hip, ‘extreme’ CT (i.e. high resolution hr pQCT) of the radius or tibia (See Figure 1), TBS (i.e. trabecular bone score), histomorphometry and magnetic resonance imaging of the radius (22-26). However, more work still needs to be done to ascertain their role in clinical medicine. Still, BMD represents the most accurate, cost effective, and easiest parameter for risk assessment (21). In part, two-dimensional DEXA measurements integrate actual bone mineral content in both trabecular and cortical compartments with bone size. Due to the strong association between bone mineral density and future fractures, this phenotype remains an excellent surrogate for defining both the genetic and acquired components of the disease process. TBS, (trabecular bone score) in the vertebrae, which can be measured from DXA, provides a relatively rapid way of assessing bone quality, although its role in providing additional risk assessment remains to be determined (24).
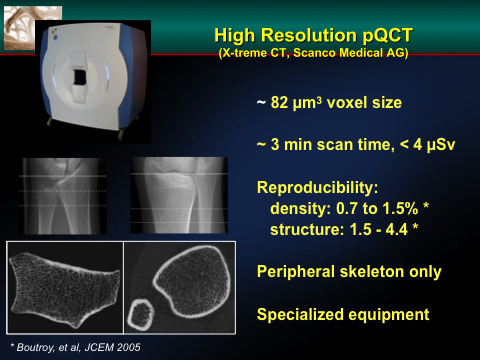
Figure 1.
This is a high resolution QCT image of the distal radius of an individual patient. One can image the trabecular bone of the peripheral skeleton and define measures of bone “quality” by specific measurements. It is still not clear whether these measurements provide a better insight into fracture risk than DXA.
BONE REMODELING AND ITS RELATIONSHIP TO BONE QUANTITY
Adult bone mineral density represents the end result of two processes; acquisition of peak bone mass during adolescence and maintenance of bone mass during the middle and later years. Changes in bone mass result from physiologic and pathophysiologic processes in the bone remodeling cycle (25). This can occur during the stage of accelerated linear growth in adolescence, or much later in life, usually after menopause in women. The bone remodeling cycle is a tightly coupled process whereby bone is resorbed at approximately the same rate as new bone is formed. Basic multicellular units (BMUs) compose the remodeling unit of bone and include: osteoclasts which stimulate bone resorption, osteoblasts, which are responsible for new bone formation, and osteocytes, older osteoblasts surrounded by bone and present in a reduced state of activity (26) (See Figure 2). Activation of the remodeling cycle serves two functions in the adult skeleton: 1) to produce a supply rapidly, as well as chronically, of calcium to the extracellular space; 2) to provide elasticity and strength to the skeleton. When the remodeling process is uncoupled so that resorption exceeds formation, bone is lost. On the other hand, during peak bone acquisition, formation exceeds resorption resulting in a net gain of bone. Remodeling is more pronounced in the trabecular skeleton (e.g. spine, calcaneus and proximal femur) due to much greater surface area, and is the most metabolically active component of bone, in part because of its proximity to the marrow space. However, trabecular bone is also extremely vulnerable to perturbations by local or systemic factors that can cause significant imbalances in bone turnover.
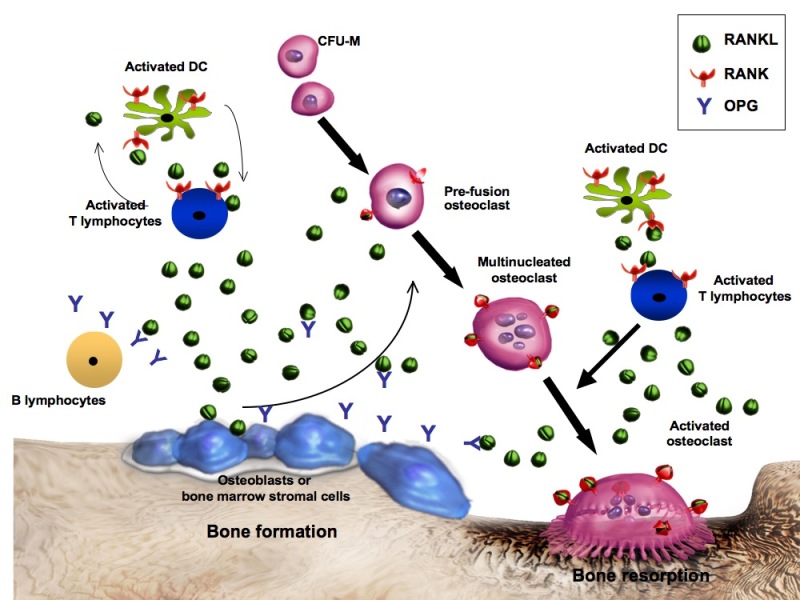
Figure 2.
The bone remodeling cycle. The osteoblast (OB) orchestrates the orderly process of bone remodeling through activation signals from systemic factors including growth hormone (GH), interleukins (IL-1,IL-6), parathyroid hormone (PTH), and withdrawal of estrogen (-E2). M-CSF and RANKL are the two major OB mediated factors which regulate the recruitment and differentiation of the osteoclast (OC). Osteoprotogerin (OPG) is also synthesized by OBs and serves as a soluble decoy receptor blocking activation of RANK. Inhibition or knockout of these signals from OB-OC results in reduction in bone resorption. Other cells including activated T lymphocytes may contribute to the marrow milieu. Not pictured are IGFs which are released during bone resorption and serve as coupling factors to recruit new OBs to the surface. These peptides may also be important for osteoclast activity. Also sclerostin, a peptide produced by osteocytes reduces new bone formation by blocking Wnt signaling.
The initiation of the bone remodeling cycle remains unclear. The long-standing dogma was that activation of resting osteoblasts on the surface of bone and marrow stromal cells began the process (27-33). This would mean that there were initiating signals, either paracrine or endocrine that would stimulate osteogenesis. One possibility is that osteocytes, which can sense fluid shifts and microcracks, and are imbedded deep within the matrix of the skeleton, induce the remodeling sequence, by paracrine signaling to the osteoblast (27-29). But those same cells secrete factors that can initiate osteoclast differentiation as well as alter osteoblast mediated bone formation. Osteocytes, for those reasons, are now considered the ‘command and control’ system for remodeling. More recently, investigators believe that RANKL, released from osteocytes as well as stromal cells, drives osteoclast differentiation, beginning the process of active resorption prior to osteoblast differentiation. This can occur as a result of hormonal signals or mechanical loading. Resting lining cells or osteoblast progenitors as well as mesenchymal stromal cells can become activated at the same time when osteoclast differentiation is ongoing. Those progenitors also signal to osteoclast precursors to induce further differentiation and induction of hematopoietic cells (29). Osteoclasts, once differentiated, may also elaborate growth factors, such as the Wnts and sphingosine to signal back to osteoblast progenitors. After osteoclast- induced bone resorption, matrix components such as TGF-beta and IGF-I, as well as collagen, osteocalcin, and other protein and mineral components, are released into the micro environment of the niche. Growth factors released by resorption contribute to the recruitment of new osteoblasts to the bone surface, which begin the process of collagen synthesis and mineralization. But, in addition stromal factors such as the Wnts can induce further osteoblast differentiation via the LRP5/6 signaling pathway. In healthy adults as many as two million remodeling sites may be active at any given time, and it is estimated that nearly one fourth of all trabecular bone is remodeled each year. In general resorption takes only 10-13 days, while formation is much more deliberate and can take upwards of three months (Figure 2). Under ideal circumstances, by the end of the cycle, the amount of bone resorbed equals the amount reformed. Cessation of bone formation almost certainly occurs via osteocyte mediated sclerostin (see below) which blocks further Wnt signaling. In sum, remodeling begins at the surface of the trabecular and cortical bone via signals from the osteocytes, probably starting with osteoclast differentiation and then signaling backwards to osteoblasts, and vice versa.
In contrast to normal remodeling, osteoporosis has been classically defined in a pathogenic manner as an uncoupling in which resorption exceeds formation resulting in a net loss of bone. However, it is also apparent that some individuals have impaired peak bone acquisition. This scenario may be more common than previously appreciated and almost certainly represents inherited or acquired alterations in the rate of either bone formation or bone resorption during a critical period when several hormones in synchrony orchestrate a marked increase in bone mass (see later).
There are several key components of the remodeling cycle which are susceptible to systemic and local alterations and when perturbed, can lead to deleterious changes in bone mass. In particular, activation of remodeling via the osteoblast, and recruitment of osteoclasts, represent the two most vulnerable sites in the cycle. A third cell, altered by disease states is the osteocyte, an entombed fully differentiated osteoblast that connects to the surface osteoblasts, and likely senses mechanical stimulation. Osteocyte apoptosis may contribute to age-related osteoporosis either directly or through the elaboration of systemic peptides. Interestingly, remodeling may end with the osteocyte as well, since it produces a protein, sclerostin, which inhibits osteoblast activity by antagonizing the Wnt and BMP pathways (see below) (34). Monoclonal antibodies that bind to sclerostin have been developed and one (e.g. romosozumab) has been approved for the treatment of postmenopausal osteoporosis (35-36). That monoclonal antibody enhanced bone formation, increased bone mineral density by 13-15% at one year, suppressed bone resorption (via Wnt mediated RANKL), and reduced overall fractures of the spine (32). Finally, one could consider macrophages to have an important role in remodeling, since these cells are present in the bone marrow niche and respond to injury with inflammatory cytokines and immune modulators (37). On the other hand, the bone marrow niche may be protected against macrophage induced cytokine release due to a protective ‘canopy’ of lining cells, although recent evidence suggests that there is a pro-inflammatory response in the marrow with certain perturbations such as diet induced obesity or aging (29).
Uncoupled remodeling occurs during menopause, with estrogen deprivation or antagonists, or in response to endogenous parathyroid hormone fluxes, cytokine stimulation, growth hormone surges, glucocorticoid excess, or changes in serum calcium. For the most part, estrogen deprivation remains one of the most common and critical elements in shifting resorption rates to a higher set point (38-42). Although bone formation initially can "catch up", the length of time for each component of the remodeling cycle clearly favor resorption over formation as the process of laying down new bone requires the interaction of several processes (see Figure 2). But, it is still unclear why falling estrogen levels, which is a universal event during the menopausal years, causes such rapid bone loss in a relatively small percentage of women (43). Clearly, factors such as peripheral conversion of testosterone to estradiol, adrenal androgen production, FSH levels, and genetic determinants, as well as other local signals may also be important. In regards to FSH, SWAN (Studies of Women Across the Ages) reported that higher FSH precedes estrogen loss and this is associated with longitudinal bone loss more closely than circulating estrogen. Although not identified in humans, mice have strong heritable determinants that affect the rate of age-related bone loss and in one mouse model high FSH levels drove significant bone loss independent of estrogen.
The nature of the osteocyte-osteoblast-osteoclast interaction has been one of the most active areas of recent investigation (see Figure 2). External signals (such as PTH, growth hormone, interleukin-1, estrogen deprivation) to resting osteoblasts and stromal cells cause these cells to release a potpourri of cytokines (i.e. interleukins such as IL-1, -6, -11 as well as m-CSF, tumor necrosis factor (TNF), and TGF-beta) that enhance the recruitment and differentiative function of multinucleated giant cells destined to become bone resorbing cells (38). However, one of the most critical pathways in the osteoblast-osteoclast interaction scheme is the RANKL- Osteoprotogerin (OPG) relationship. OPG is a soluble peptide originally described as a factor which markedly inhibited bone resorption and osteoclast differentiation in vitro (42). This protein is a member of the TNF receptor super-family and its role in bone remodeling is to act as a decoy receptor for the peptide known as osteoprotegerin ligand i.e. OPGL (or RANKL) (42). In fact, RANKL is a surface peptide which when expressed on the osteoblast, binds to the true OPGL receptor (also called RANK-receptor activator of NFkB Ligand) on osteoclasts, and initiates cell-cell contact necessary for osteoclast activation and subsequent bone resorption (42). More recently RANKL has been shown to be produced by osteocytes and can result in osteocytic osteolysis during states of high calcium demand such as lactation, estrogen deficiency, and even acute exercise (43).
The OPG, OPGL and RANK system that affects osteoclast differentiation, in addition to the effects of m-CSF on osteoclast proliferation, provides the critical link among osteocytes, osteoclasts and osteoblasts. It has also led to the synthesis of RANKL antibodies. Denosumab, (brand name Prolia) was the first approved monoclonal antibody against RANKL for the treatment of postmenopausal osteoporosis due to its strong efficacy in reducing spine and hip fractures (44-46). It is administered once every six months and it suppresses bone resorption by 80-90%. Unlike the bisphosphonates, denosumab’s anti-resorptive effect wanes in 4-6 months, thereby providing a margin of safety in terms of total suppression in remodeling (46). On the other hand, because the anti-resorptive effect wanes rapidly, there is concern about post treatment rebound and fractures, particularly of the vertebral spine (47).
The osteoblast functions not only to signal osteoclasts during remodeling as well as receive signals from them, but also to lay down collagen and orchestrate mineralization of previously resorbed lacunae in the skeletal matrix. These complex functions are tied to differentiation of mesenchymal stromal cells which become terminally differentiated osteoblasts and rest on the surface of the remodeling space (29). Recruitment of stromal cells or lining cells into osteoblasts, rather than adipocytes is a critical step in bone formation and requires a series of transcription factors that enhance differentiation. This is particularly important since one of the features of estrogen deficiency and age-related osteoporosis is the development of increased bone marrow adiposity (48,49). Indeed, the presence of excess bone marrow fat may be a major risk factor for osteoporotic fractures (50).
One of the most important components driving osteogenesis is Runx2, a unique transcription factor which is essential in the early differentiation pathway of osteoprogenitors (51). Regulation of Runx2 has become a major focus of work in as investigators have begun to consider novel ways to enhance bone formation and reduce marrow adipogenesis (50). Also, it should be noted that there are metabolic programs activated by transcription factors that are essential for fueling the work of the osteoblast. Conditions of substrate insufficiency; e.g. anorexia nervosa, diabetes mellitus etc. also impair bone formation by altering their metabolic programs.
With activation of resting osteoblasts and lining cells, osteoblasts synthesize several types of collagen as well as elaborating a series of growth factors such as IGF-I, IGF-II, TGF-β. These, in turn, are necessary for further recruitment of bone forming cells (52). In addition, osteoblasts deposit growth factors in the skeletal matrix where they are stored in latent forms, and released during subsequent remodeling cycles. After deposition of new bone, some osteoblasts are encased by matrix. These osteocytes, are still viable, although less metabolic and, can, through newly developed caniculi, provide signals to other bone cells. Indeed, most evidence suggests that osteocytic signals are important in the so-called "mechanostat", the gravitary sensing device which modulates bone formation, as well as initiating normal remodeling sequences. As noted earlier, osteocytes may participate in bone resorption of the cortex by secreting RANKL. This is termed ‘osteocytic osteolysis’ and may occur during lactation and other states of high calcium demand.
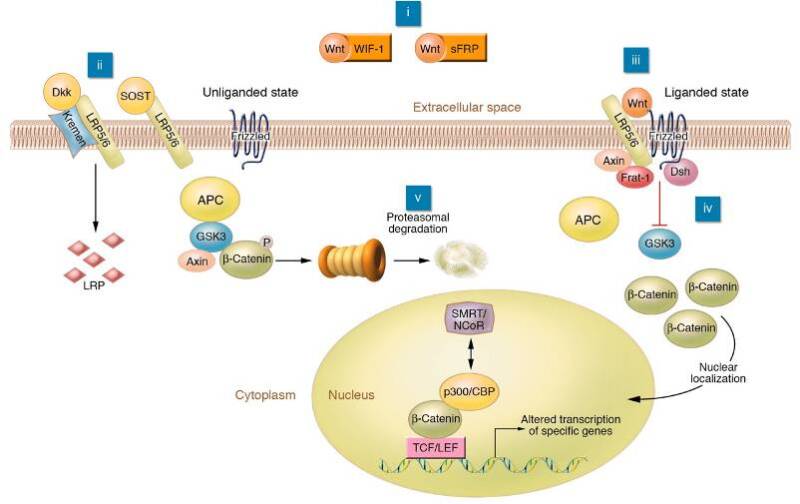
In the last half-decade, the Wnt/β catenin signaling pathway has emerged as a major regulator of bone formation and a potential mediator of the mechanostat. Wnts belong to a large family of peptides that bind to two membrane bound receptors, Lrp5 or 6 and Frizzled (see Figure 3) (53). Once these ligands bind, activation occurs through a complex intracellular signaling pathway mediated by /β catenin which translocates to the nucleus and stimulates the transcription of several genes through the TCF/Lef network. Sclerostin (SOST), the osteocyte specific protein that inhibits bone formation does so by binding to Lrp5 and blocking Wnt signaling (54). The anti-sclerostin antibody, romosozumab, was shown to markedly increase bone mass and reduce fractures in postmenopausal women after only 1 year of treatment (55,56). It was approved by the FDA and European Regulatory Agencies for the treatment of postmenopausal osteoporosis.
In summary, the bone remodeling cycle is complex and redundant. The three major cells, osteocytes, osteoblasts and osteoclasts, arise from different stem cells (mesenchymal for osteoblasts and osteocytes, and hematopoietic for osteoclasts) and are under the control of various factors which in harmony orchestrate an orderly remodeling sequence. Their birth and death (i.e. the cycle of recruitment, proliferation and programmed cell death) and the regulatory factors which control those events, are also complex, yet vitally important for understanding the pathogenesis of osteoporosis. Alterations at any stage along the process of recruitment, activation, differentiation, or cell death can lead to imbalances in remodeling which eventually would result in bone loss, reduced bone mass and ultimately fractures. Some of those perturbations are noted below.
SYSTEMIC AND LOCAL ALTERATIONS IN THE BONE REMODELING SEQUENCE LEAD TO BONE LOSS
The importance of estrogen in maintaining calcium homeostasis for the postmenopausal woman was first established by Fuller Albright, more than 70 years ago (57). Since that time more evidence has accumulated from randomized intervention trials demonstrating that hormone replacement (estrogen with or without progesterone) reduces bone turnover and increases bone mass (58). However, these data provide only indirect evidence that estrogen levels are important as pathogenic components of the osteoporosis syndrome. More recent studies provide stronger evidence of the association between low estradiol concentrations and low bone mass. Several investigators have demonstrated that the lowest estradiol levels in postmenopausal women (i.e. <5 pg/ml) are associated with the lowest bone mineral density and the greatest likelihood of fracture (59). In addition, at least one study has shown that males with osteoporosis have lower serum levels of estradiol then do age-matched men who do not have low bone mass (60). Moreover, there are now two case reports describing mutations in either aromatase activity or the estrogen receptor, which produced a phenotype of severe osteoporosis in men (61,62). In the former case, estrogen replacement therapy for this young man, resulted in a marked increase in spine and hip bone mineral density. In both situations the lack of functional estrogen, despite normal to high levels of testosterone, resulted in very low BMD (63).
Although declining estradiol levels contribute to the osteoporosis syndrome, the precise molecular events or sequences that result from changes in ambient hormonal concentrations are not clear. In some animal models, estrogen deprivation is associated with a marked increase in IL-6 synthesis from stromal and osteoblastic cells. This is consistent with experimental findings which demonstrate that estrogen regulates the transcriptional activity of the IL-6 promoter (39,41). However, results in other studies are conflicting. In other experimental paradigms, changes in TNF, IL-11 and IL-1 can all be associated with increased bone resorption (40). Recently RANKL has been identified as a major regulator of osteoclast differentiation. Thus, it seems likely that several cytokines, working in concert, are active during estrogen deprivation, and each can accelerate the process of bone resorption. RANKL, however may be the most critical and necessary for full activation of remodeling. Enhanced bone resorption eventually leads to bone loss from estrogen deprivation since bone formation rates cannot keep up with rates of bone resorption (31,42, 43). Moreover, the faster the resorption, the greater the loss resulting in more damage to the structure integrity of the skeleton.
In contrast to the plethora of studies on bone loss and estrogen, there are few good studies relating androgen deprivation to bone loss in women. Androgen receptors are present on osteoblasts. However, both in vitro and in vivo studies in men have yielded conflicting results. Like estrogen, androgens can regulate the IL-6 promoter and in experimental animals, orchiectomy has been associated with increased IL-6 production and bone loss (64). In men, chronically low androgen levels have been associated with low bone mass, and testosterone replacement can enhance bone mineral density (65). However, estradiol levels in men may be a more important risk factor for fracture than androgen levels and correlate more closely with trabecular bone volume as measured by QCT. At the present time it is not clear precisely what role androgens play in the maintenance of bone mass in both men and women except in deficiency syndromes, particularly hypogonadism.
Other more common causes of male osteoporosis include alcoholism, glucocorticoid excess, and hypercalciuria (see Table 2). In the first two cases, hypogonadism remains a pathogenic feature of that osteoporosis syndrome. Less frequently, men with gluten enteropathy or an endocrinopathy such as thyrotoxicosis or primary hyperparathyroidism can present with multiple fractures and low bone mass (see Table 2).
Table 2.
Etiology of Osteoporosis in Men
| Etiology | Age-years | Clinical Features |
|---|---|---|
| Hypogonadism | 30-80 | low Test, low E2, increased resorption |
| Alcoholism | 40-80 | low test, E2+/-, +/- turnover |
| Glucocorticoids | 20-80 | +/- test, E2 +/-, increased resorption, Decreased formation |
| Multiple myeloma | 40-80 | Hypercalcemia, ESR |
| Hypercalcuria | 30-80 | Test, E2 nl; increased resorption Hypercalcuria, inc PTH, kidney stones |
| Idiopathic Osteoporosis- | 40-80 | fractures, low formation, low IGF-I |
| Gluten enteropathy | 20-80 | low 25OHD, turnover increased |
| Endocrine Disorders | 15-90 | Cushing’s ± |
| PHPT | 15-90 | PTH increased in all cases; increased resorption |
| Thyrotoxicosis | 15-90 | Decreased PTH |
E2- estradiol; Test-testosterone; PTH-parathyroid hormone; PHPT-primary hyperparathyroidism; Inc- increased
In women, bone loss is accelerated immediately after menopause. However, recent studies demonstrate that markers of bone resorption are also very high later in life. In particular, women in their 80s and 90s have been noted to lose bone at a rate of greater than 1% per year from the spine and hip (65), Contrary to earlier studies, it is now evident that the older woman who is not as physically active, and is not on estrogen, is at extremely high risk of bone loss and subsequent fractures. The pathogenesis of this process is multifactorial although dietary calcium deficiency, leading to secondary hyperparathyroidism, certainly plays a central role. The average calcium intake of women in their 8th and 9th decades of life is now estimated to be between 800-1000 mg/day (66). If vitamin D intake is also sub-optimal, and serum levels of 25 OH vitamin D <20 ng/ml, or 50nmol/l, secondary hyperparathyroidism may occur, although there are other causes for increases in PTH, including chronic renal insufficiency and low calcium diet (67). PTH stimulates osteoblasts and provokes the remodeling sequence including the elaboration of several cytokines that accelerate bone resorption. Unfortunately, in most elders, bone formation is not enhanced although the reasons for this are not entirely clear beyond the simplistic notion that stem cell recruitment is impaired in the elderly. Nonetheless, this leads to further uncoupling in the bone remodeling cycle, and significant bone loss. Among elders with poor calcium intake who live in northern latitudes, seasonal changes in vitamin D lowering levels below 20 ng/ml might aggravate bone loss (68,69). Whether increased bone loss from vitamin D deficiency is an independent risk factor for future fractures in the elderly remains somewhat controversial necessitating further studies to define such a risk. Overall, there is evidence that vitamin D deficiency is associated with a greater risk of fracture in frail elderly institutionalized men and women (<20 ng/ml; 50 nmol/l), whereas it is unlikely that those individuals with 25 OHD levels between 20-30 ng/ml are at a greater risk (70). The recently completed VITAL trial demonstrated that 2000 IU of vitamin D to healthy men and women did not prevent bone loss or alter bone turnover (LeBoff, personal communication and abstract ASBMR,2019).
There is growing evidence that low serum levels of vitamin D, through impaired calcium absorption, can stimulate PTH release and increase bone turnover in the elderly. Increased PTH enhances 1,25 dihydroxyvitamin D, and this in turn could suppress further bone formation and mineralization (71). Thus, bone loss is associated with uncoupled remodeling. Many older individuals already have established osteoporosis. Coincidental vitamin D deficiency due to poor intake, absent sunlight exposure, or impaired conversion of vitamin D to its active metabolite, can result in osteomalacia as well as aggravating pre-existent osteoporosis (70). LeBoff et al reported that more than 50% of elders who presented with a hip fracture were frankly vitamin D deficient (71). Therefore, the combination of vitamin D deficiency with inadequate calcium intake enhances the likelihood of rapid bone loss in the very susceptible elderly population. Still, it is unclear how secondary hyperparathyroidism causes bone loss. Chronic elevations in PTH secretion due to primary or tertiary hyperparathyroidism, have been associated with low bone mass at several skeletal sites including the radius. Elevated PTH levels in older women have been associated with bone loss in some studies but not in others. In elderly individuals, it has been reported that PTH levels are closely correlated with increased synthesis of an insulin-like growth factor binding protein (IGFBP-4) which suppresses IGF action on bone cells and may increase sclerostin secretion (72, 34). Since IGF-I is an important growth factor for osteoblasts, it is conceivable that PTH down regulates IGF activity during states of relative calcium/and or vitamin D deficiency. This would shift the remodeling balance towards preserving intravascular calcium concentrations, while inhibiting new calcium incorporation into the skeletal matrix. In sum, there is little doubt that calcium and vitamin D insufficiency are prominent causes of accelerated bone loss in the elderly (73). However, in the healthy postmenopausal population, there is little evidence that vitamin D supplementation prevents bone loss or fractures.
As noted previously, high circulating levels of glucocorticoids have a significant impact on bone acquisition and maintenance. In 1932 Harvey Cushing recognized the syndrome of endogenous steroid excess which included marked osteopenia and fractures (74). Long term exposure to pharmacologic doses of glucocorticoids results in significant bone loss and enhanced marrow adipogenesis as marrow stromal cells differentiate down the fat lineage. In addition to having direct effects on the osteoclast and osteoblast, glucocorticoids also induce secondary hypogonadism and hyperparathyroidism, impaired vitamin D metabolism, muscle atrophy, and hypercalciuria (See Table 3). All these factors contribute to a rapid and sustained loss of bone during the first few months of steroid therapy (75). The addition of other immunosuppressants such as cyclosporine has been shown to aggravate bone loss by further increasing bone resorption. As the number of organ transplants have increased exponentially over the last decade, the prevalence of post-transplantation osteoporosis has risen substantially. Steroid induced osteoporosis is now considered the second most common cause of low bone mass in the general population and one of the most common causes of osteoporotic fractures (75). It is listed in the FRAX data set as one critical risk factor to assess in determining 10-year fracture likelihood.
Table 3.
Effects of Glucocorticoids on Bone Mass
| Response to Glucocorticoids | Effects on Bone Remodeling | Effects on Bone Mass |
|---|---|---|
| Increased PTH secretion | Increased bone resorption ? decreased bone formation | rapid loss of bone |
| Decreased LH/FSH secretion | Increased bone resorption due loss of estrogen | loss of bone |
| Impaired calcium absorption due to decreased 1,25 D | Increased PTH, increased bone resorption | loss of bone |
| Increased calcium loss in urine | Secondary increase in PTH, Increased bone resorption | loss of bone |
| Acute suppression of osteoblasts and apoptosis | Reduced bone formation | gradual bone loss |
| Stimulation of osteoclastogenesis | Increased bone resorption rapid | loss of bone |
PATHOGENIC FACTORS WHICH IMPAIR PEAK BONE MASS
Peak bone mass is acquired between the ages of 10-16 years. It is the zenith of bone acquisition and represents the sum of several processes including a marked increase in bone formation (76,77). Boys tend to reach peak 2 years later than girls and their bone mineral density is higher than women at all skeletal sites. In part this relates to a greater cross-sectional bone area in males than females (78). Peak bone mass results from linear growth and consolidation of cortical and trabecular components. Acquisition is most rapid during the latter stages of puberty and coincides with maximum growth hormone secretion, high serum IGF-I levels, and rising levels of estradiol and testosterone. In addition, calcium absorption is maximal and skeletal accretion is optimal. All these processes combine over a relatively short period of time to produce a bone mass that subsequently plateaus and then falls during later life. It is estimated that more than 60% of adult bone mass can be related to peak acquisition. Hence understanding the mechanisms responsible for low bone mass must include perturbations in peak bone acquisition.
There are several hormonal, environmental, nutritional and heritable determinants of peak bone mass. These include estrogen/testosterone, growth hormone/IGF-I, adequate nutrition, calcium/vitamin D, and unknown genetic factors. If each is perturbed, dramatic alterations in peak bone mass may occur, setting the stage for low bone density throughout life. Gonadal steroids are important not only to bone maintenance but also to its acquisition. During puberty, estrogen and testosterone levels rise and contribute to consolidation of bone mass. Estrogen is also necessary for epiphysial closure. Studies of a male with an estrogen receptor mutation and men with an aromatase deficiency established that estradiol is critical for bone acquisition (61,62). These young men share several phenotypic characteristics including tall stature unfused epiphysis, and very low bone mass. Hence, there must be a threshold effect for estradiol in men, and this effect must be time dependent. Similar conclusions can be drawn from studies in women. Acquired deficiencies in estrogen, such as anorexia nervosa, or chemotherapy induced ovarian dysfunction, result in low peak bone mass and lead to subsequent risk for osteoporosis (79,80,81). Nearly identical findings have been noted in patients with untreated Turner's syndrome and in men with Klinefelter's syndrome, although the high FSH levels could also contribute to bone loss.
The timing of gonadal steroid surges is critical for bone acquisition since there is a relatively short window of time in which bone formation is favored and matrix synthesis is markedly enhanced. That window is likely to be less than three years and earlier in girls than boys. Probably the best study which addressed this issue comes from a retrospective analysis of men in their thirties who underwent late onset of puberty (i.e. at the age of 17 or 18) but were otherwise normal by full endocrine testing. These men had significantly lower bone mineral density in their thirties than age matched men who went through puberty at a normal time. These data suggest that timing as well as quantity of gonadal steroids is critical for bone acquisition.
Pubertal surges of estrogen and androgens are also important for priming the growth hormone/ IGF-I axis. Rising levels of both contribute to growth hormone surges that lead to increases in circulating and tissue expression of IGF-I, an essential growth factor for chondrocyte hypertrophy and expansion. IGF-I may also be critical in defining the cross-sectional size of bone, a potentially important determinant of bone strength. Once again, studies in growth hormone deficient, or growth hormone resistant individuals have established that low levels of circulating IGF-I, especially during puberty, are associated with reduced bone mass (82). In addition, rhGH replacement has been shown to restore linear growth and improve peak bone mass acquisition. Several studies in experimental animals, including inbred strains of mice, have established that IGF-I is important for bone acquisition and the timing of IGF-I peaks coincide with maximal rates of bone formation. Impairment in production of IGF-I due to acquired disorders such as anorexia nervosa, malnutrition, and delayed puberty can also impede peak bone acquisition (80). Recently, it has become apparent that Type I IDDM can impact the bone marrow niche and suppress bone formation and increase resorption. This uncoupling can lead to an impairment in peak bone mass, although there are other determinants of glucose intolerance that can impact the skeleton; e.g. increased advanced glycation end products, glucose toxicity, material property changes in the matrix.
Hormonal abnormalities not only enhance bone resorption in older individuals, but may blunt the capacity of bone cells to maximize bone formation during adolescence. Clearly, hypogonadal boys and girls have impaired peak bone mass, resulting in low adult bone mineral density. Even one form of contraception, Depo-provera, may reduce estrogen concentrations enough in the teen girl, to reduce her capacity to acquire peak bone mass. Similarly, it seems likely although not proven that smoking during the teen years could impair osteoblast activity and flatten projected trajectories for peak bone acquisition.
More recently diabetes mellitus has been established as a secondary cause of osteoporosis. TIDM, particularly at early onset (e.g. childhood or adolescence) is associated with low bone mass and fractures (83). Both hip and vertebral fractures are more common in adult T1DM, whereas in T2D, in which areal BMD is normal, fracture risk is increased by 20%, but the type of fractures are usually peripheral in nature (84). Importantly use of rosiglitazone, a TZD is also associated with an increased risk of fracture whereas DPP4 inhibitors and GLP1 agonists are bone neutral or favor a slight increase in bone mass. The newer SGLT-2 inhibitors are associated with a greater risk of fracture as a class. Despite the greater risk with glucose intolerance, the pathophysiology of these disorders is not well defined, although changes in advanced glycation end products, enhanced reactive oxygen species, and glucose toxicity are all considered likely contributors.
In order to mineralize newly synthesized bone, calcium must become bioavailable to the skeletal matrix. In experimental studies in rodents and humans, it is clear that the several pools of available calcium are markedly enhanced during puberty. These include calcium efflux from the gastrointestinal tract, and the calcium pool available for incorporation in the matrix. It is no coincidence that growth hormone surges not only increases IGF-I, (thereby enhancing skeletal growth and matrix biosynthesis) but also result in increases in 1,25 dihydroxyvitamin D (possibly via IGF-I induction of 1, alpha hydroxylase activity), the active metabolite of vitamin D which markedly enhances calcium absorption from the gut. Although there are no longitudinal studies in pubertal individuals with prolonged calcium deficiency, several randomized placebo-controlled trials in pubertal and pre-pubertal girls and boys have established that supplemental calcium can enhance bone mineral density. In a twin study, in which one twin receives 1200 mg of calcium supplementation, and one receives placebo, radial BMD increases by as much as 5% after three years compared to placebo (81). This study suggests that there is significant gene-environmental interaction, and that even in those individuals with heritable determinants of low peak bone mass, calcium supplementation may provide an important and relatively simple means of protecting individuals from future osteoporotic fractures.
GENETIC DETERMINANTS OF PEAK BONE MASS
Probably the most important determinant of peak bone mass, but one that has lacked clear definition is the genetic contribution. In part this is due to the complex nature of bone mineral density as a trait. Nevertheless, low peak bone mass may be the most important pathogenic factor in the osteoporosis syndrome of later life. And, it appears that at least 50% of peak bone mass is determined by genetic factors (85). What are these determinants and how are they modified by environmental factors?
Efforts to define heritable determinants of peak bone mass have been complicated by a number of issues which are also common to analyses of other complex traits. These include the following:
- 1.
A quantifiable phenotype;
- 2.
Heterogeneity within a given population under study;
- 3.
The polygenic nature of the disorder;
- 4.
Epistasis (gene-gene interaction)
- 5.
Pleiotropy- phenotypic differences with identical genotypes, and
- 6.
Gene by environmental interactions.
Notwithstanding these complexities it is clear that BMD is an acceptable phenotype for defining heritable determinants of future risk. Bone mineral density in the population is distributed in a gaussian manner, thereby allowing analyses at the extremes (<-2.0 SD or > 2.0 SD) of the density distribution. Large homogeneous and heterogeneous populations are now being studied as part of the GEFOS consortium, to ascertain genetic determinants of BMD and fractures in humans (86-91). Candidate genes are those associated previously by biologic determinants or previous studies as being important for skeletal maintenance. Indeed, many of these have been identified by whole genome studies (GWAS) include RANKL, OPG, the vitamin D receptor, collagen IA1, the estrogen receptor, interleukin-1, and IGF-I. Additionally several other candidate genes that have not been previously associated with osteoporosis have been identified. These studies have been reviewed in depth elsewhere (86-89). Depending on the cohort, the phenotype, and the number of individuals studied, it is predicted there will be hundreds of genes that contribute to individual variation in bone mass (90,91). Twin studies examining discordant or concordant phenotypes are also helpful, as are sib-pair studies, although the results have been less generalizable.
One example of a candidate gene that has also been shown to have strong heritability and predictive value for osteoporosis from GWAS studies is Lrp5 (92). Originally Recker and colleagues identified an extended family with very high bone density and fine mapped the locus to a region in chromosome 11. After several years of intense high through put analysis, that group identified a 'high bone density' gene, LDL receptor related protein 5 (LRP-5) that was mutated in this family (92). The low affinity lipoprotein receptor is important in binding Wnts, ligands critical for cell differentiation in several organisms. At the same time, Warman and colleagues identified several children with osteoporosis pseudoganglioma syndrome and subsequently mapped the gene which resulted in 'loss of function' in these individuals (93). This turned out to also be Lrp5. The potential pathways that direct osteoblast function and mineralization through Lrp5, and its co-receptor frizzled, have opened up new areas of investigation (See Figure 3 and discussion above). Moreover, natural antagonists to the Wnt/Lrp5 signaling system including sclerostin and DKK-1 have been studied using genetic engineering in mice. This pathway continues to show great promise for therapeutic interventions such as romosozumab, recently approved by the FDA for the treatment of postmenopausal osteoporosis (54, 99).
In the past five years, Lrp5 has been studied extensively both in its function and its allelic effects through genome wide association studies as well as in translational bench work. Hence this pathway is important in regulating peak bone mass. But it is also clear that since BMD is a polygenic trait, other genes are now being discovered. Moreover, gene environmental interactions must play a major role in defining heritable risk for low BMD and fracture. In addition to the search for osteoporosis genes, intervention studies in adolescents have provided insight into at least one of the environmental impacts on genetic determinants (94-97). A twin study in Indiana revealed that as long as calcium supplementation continued during puberty, young boys could enhance their peak bone mass. In a Swiss study, younger pre-pubertal girls supplemented with a protein product had a significant increase in spine bone density, as did a cohort of pubertal girls receiving a milk powder in England (96,97). Remarkably, in the latter cohort, serum IGF-I levels also rose dramatically, providing further indirect evidence of a link between pubertal status, bone mass, and the growth hormone/IGF-I axis. Thus, there is strong evidence that nutritional, hormonal and environmental factors play a major role in regulating peak bone mass.
SUMMARY
The epidemiology of osteoporosis is well established and risk factors have been defined. On the other hand, the pathogenesis of osteoporosis is complex and multifactorial (41,98). Alterations in bone mineral density almost certainly represent the final common pathway by which pathologic factors affect risk of future osteoporotic fracture. The interplay of various physiologic processes which result in peak bone mass, and maintenance of adult bone mass are key to understanding the pathogenesis of this disease. Changes in hormonal status, and in particular estradiol, clearly are important factors in regulating both bone formation and bone resorption in men and women. Perturbations in growth hormone activity, musculoskeletal function, dietary intake of calcium and vitamin D, and genetic determinants are also important pathogenic factors. Defining the role of genetic factors and their interaction with many of the environmental and hormonal determinants that have been established as potential etiologic agents responsible for low bone mineral density and fracture will certainly be the most difficult challenge facing basic and clinical researchers. On, the other hand, the strength of data from basic and clinical studies over the last decade, now allows practitioners to confidently diagnose osteoporosis. New treatment strategies offer greater hope for patients suffering from this disease.
REFERENCES
- 1.
- Cummings SR, Nevitt MC, Browner WS, Stone K, Fox KM, Ensrud KE, Cauley J, Black D, Vogt TH. Risk factors for hip fracture in white women. N Engl J Med. 1995;332:767–773. [PubMed: 7862179]
- 2.
- Kanis JA, Melton LJ, Christiansen C, Johnston CC, Khaltaev N. The diagnosis of osteoporosis. J Bone Min Res. 1994;9:1137–1141. [PubMed: 7976495]
- 3.
- Melton LJ, Chrischilles EA, Cooper C. Perspective: How many women have osteoporosis? J Bone Min Res. 1992;7:1005–1010. [PubMed: 1414493]
- 4.
- Seeley DG, Browner WS, Nevitt MC. Which fractures are associated with low appendicular bone mass in elderly women? The Study of Osteoporotic Fractures. Ann Intern Med. 1991;115:837–842. [PubMed: 1952469]
- 5.
- Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. Osteoporosis Int. 1994;4:368–371. [PubMed: 7696835]
- 6.
- Eliffors I, Allander E, Kanis JAS. The variable incidence of hip fracture in southern Europe: The Medos Study. Osteoporosis Int. 1994;4:253–261. [PubMed: 7812073]
- 7.
- Cooper C, Campion G, Melton LJ. Hip fractures in the elderly: a world wide projection. Osteoporosis Int. 1992;2:285–289. [PubMed: 1421796]
- 8.
- European Commission: Report on Osteoporosis in the European Community-Action for Prevention. Brussels; European Commission 1998.
- 9.
- DeLaet CEDH, Pols HAP. Fractures in the elderly: epidemiology and demography. Bailliere's Clin Endocrinol Metab. 2000;14:171–179. [PubMed: 11035900]
- 10.
- Ross PD. Clinical Consequences of vertebral fractures. Am J Med. 1997;103:425–435. [PubMed: 9302895]
- 11.
- Cosman F, Krege JH, Looker AC, Schousboe JT, Fan B, Sarafrazi Isfahani N, Shepherd JA, Krohn KD, Steiger P, Wilson KE, Genant HK. Spine fracture prevalence in a nationally representative sample of US women and men aged ≥40 years: results from the National Health and Nutrition Examination Survey (NHANES) 2013-2014. Osteoporosis Int. 2017;28(6):1857–1866. [PMC free article: PMC7422504] [PubMed: 28175980]
- 12.
- O Neill TW. Felsenberg D, Varlow J. The prevalence of vertebral deformity in European men and women: the European Vertebral Osteoporosis Study. J Bone Min Res. 1996;11:1010–1018. [PubMed: 8797123]
- 13.
- Cauley JA, Cawthon PM, Peters KE, Cummings SR, Ensrud KE, Bauer DC, Taylor BC, Shikany JM, Hoffman AR, Lane NE, Kado DM, Stefanick ML, Orwoll ES. Osteoporotic Fractures in Men (MrOS) Study Research Group. J Bone Miner Res. 2016 Oct;31(10):1810–1819. Risk Factors for Hip Fracture in Older Men: The Osteoporotic Fractures in Men Study (MrOS). [PMC free article: PMC5240502] [PubMed: 26988112] [CrossRef]
- 14.
- Lindsay BR, Olufade T, Bauer J, Babrowicz J, Hahn R. Arch Osteoporosis. 2016;11:19. Patient-reported barriers to osteoporosis therapy. [PMC free article: PMC4851700] [PubMed: 27129487] [CrossRef]
- 15.
- Khan M, Cheung AM, Khan AA. Endocrinol Metab Clin North Am. 2017 Mar;46(1):181–192. Drug-Related Adverse Events of Osteoporosis Therapy. [PubMed: 28131131]
- 16.
- Adler RA. Curr Opin Endocrinol Diabetes Obes. 2016 Dec;23(6):430–434. Bisphosphonates and atypical femoral fractures. [PubMed: 27653003]
- 17.
- Whittier X, Saag KG. Rheum Dis Clin North Am. 2016 Feb;42(1):177–89. Glucocorticoid-induced Osteoporosis. [PubMed: 26611558]
- 18.
- Marshall D, Johnell O, Wedel H. Meta-analysis of how well measures on bone mineral density predict occurrence of osteoporotic fractures. Br Med J. 1996;312:1254–1259. [PMC free article: PMC2351094] [PubMed: 8634613]
- 19.
- Stewart A, Walker LG, Potter RW, Primrose WR, Reid DM. Prediction of a second hip fracture: the potential role of DXA, ultrasound, and other risk factors for targeting of preventative therapy. J Clin Densitometry. 1999;2(4):363–370. [PubMed: 10677789]
- 20.
- Tinetti ME, Speechely M, Gunter SF. Risk factors for falls among elderly persons living in the community. N Engl J Med. 1988;319:1701–1707. [PubMed: 3205267]
- 21.
- Miller PD. Guidelines for the clinical utilization of bone mass measurements in the adult population. Calcif Tiss Int. 1995;57:252–252. [PubMed: 8673860]
- 22.
- Marques A, Ferreira RJ, Santos E, Loza E, Carmona L, da Silva JA. Ann Rheum Dis. 2015 Nov;74(11):1958–67. The accuracy of osteoporotic fracture risk prediction tools: a systematic review and meta-analysis. [PubMed: 26248637]
- 23.
- Riggs BL, Wahner HW, Seeman E. Changes in bone mineral density of the proximal femur and spine with ang: differences between the postmenopausal and senile osteoporosis syndromes. J Clin Invest. 1982;70:716–723. [PMC free article: PMC370279] [PubMed: 7119111]
- 24.
- Boutroy S, Bouxsein ML, Munoz F, Delmas PD. In vivo assessment of trabecular bone architecture by high resolution peripheral quantitative CT. J Clin Endocrinol Metab. 2005;90:6508–6515. [PubMed: 16189253]
- 25.
- Martineau P, Leslie WD. Bone. 2017 Nov;104:66–72. Trabecular bone score (TBS): Method and applications. [PubMed: 28159710]
- 26.
- Manhard MK, Nyman JS, Does MD. Transl Res. 2017 Mar;181:1–14. Advances in imaging approaches to fracture risk evaluation. [PMC free article: PMC5357194] [PubMed: 27816505]
- 27.
- Lian JB and Stein GS. The Cells of Bone in Principles of Bone and Cartilage Metabolism. Ed by Seibel MJ, Robbins S and Bilezikian JP. Academic Press, San Diego 1999 pp 165-185.
- 28.
- Bonewald LF. Endocrinol Metab Clin North Am. 2017 Mar;46(1):1–18. The Role of the Osteocyte in Bone and Nonbone Disease. [PMC free article: PMC5300041] [PubMed: 28131126]
- 29.
- Reagan MR, Rosen CJ. Nat Rev Rheumatol. 2016 Mar;12(3):154–68. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. [PMC free article: PMC4947935] [PubMed: 26607387]
- 30.
- Lorenzo JA and Raisz LG. Cytokines and Prostaglandins in Principles of Bone and Cartilage Metabolism Ed by Seibel MJ, Robbins S and Bilezikian JP. Academic Press San Diego CA 2005 pp 97-109.
- 31.
- Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, Nakagawa N, et al. Osteoblasts/Stromal Cells Stimulate Osteoclast Differentiation factor/RANKL but not Macrophage Colony Stimulating Factor. Bone. 1999;25:517–523. [PubMed: 10574571]
- 32.
- Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, et al. Osteoprotegrin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. [PubMed: 9603945]
- 33.
- Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocrine Rev. 1992;12:66–80. [PubMed: 1555533]
- 34.
- Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S. Bone density ligand, sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–1749. [PubMed: 17002572]
- 35.
- Xiadong L, Ominsky MS, Warmington KS, Marony S, Gong J, Cao J, Yongming G, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Zhaopo G, Qing-Tian N, Hua Zhu K, Kosteniuk PT, Scott Simont W, Lacey DL, Paszty C. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–588. [PubMed: 19049336]
- 36.
- Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, Hofbauer LC, Lau E, Lewiecki EM, Miyauchi A, Zerbini CA, Milmont CE, Chen L, Maddox J, Meisner PD, Libanati C, Grauer A. N Engl J Med. 2016 Oct 20;375(16):1532–1543. Romosozumab treatment in postmenopausal women with osteoporosis. [PubMed: 27641143]
- 37.
- Michalski MN, McCauley LK. Pharmacol Ther. 2017 Jun;174:43–54. Macrophages and skeletal Health. [PMC free article: PMC5429177] [PubMed: 28185913]
- 38.
- Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111(8):1221–1230. [PMC free article: PMC152939] [PubMed: 12697741]
- 39.
- Jilka RG, Girsole GH, Passeri G, Williams D, Abrams J, Boyce B, Broxmeyer H, Manoloagas S. Increased osteoclast development after estrogen loss: mediation by IL-6. Science. 1992;257:88–91. [PubMed: 1621100]
- 40.
- Pacifici R, Brown C, Puscheck E. The effect of surgical menopause and estrogen replacement on cytokine release from human blood monocytes. Proc Natl Acad Sci USA. 1991;88:5134–5138. [PMC free article: PMC51826] [PubMed: 2052592]
- 41.
- Manolagas SC, Jilka RL. Emerging insights into the pathophysiology of osteoporosis. N Engl J Med. 1995;332:305–311. [PubMed: 7816067]
- 42.
- Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O'Brien CA. R. J Biol Chem. 2016 Nov 25;291(48):24838–24850. ANKL (Receptor activator of NFκB ligand) produced by osteocytes Is required for the Increase in B cells and bone loss caused by estrogen deficiency in mice. [PMC free article: PMC5122756] [PubMed: 27733688]
- 43.
- Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, Bonewald L, Manolagas SC, O'Brien CA. PLoS One. 2015 Sep 22;10(9):e0138189. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. [PMC free article: PMC4578942] [PubMed: 26393791]
- 44.
- Zebaze R, Libanati C, McClung MR, Zanchetta JR, Kendler DL, Høiseth A, Wang A, Ghasem-Zadeh A, Seeman E. J Bone Miner Res. 2016 Oct;31(10):1827–1834. Denosumab reduces cortical porosity of the proximal femoral shaft in postmenopausal women with osteoporosis. [PubMed: 27082709]
- 45.
- Cummings, SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wong A, Kutilek S, Adami S, Zanchetti J, Libanati C, Siddhanti S, Christiansen C: FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 209 Aug 20:361(8),756-765. [PubMed: 19671655]
- 46.
- Ottawa (ON): Denosumab (Xgeva) [Internet]. Canadian Agency for Drugs and Technologies in Health; 2016 Nov.
- 47.
- Lamy O, Gonzalez-Rodriguez E, Stoll D, Hans D, Aubry-Rozier B. J Clin Endocrinol Metab. 2017 Feb 1;102(2):354–358. Severe rebound-associated vertebral fractures after denosumab discontinuation: nine clinical cases report. [PubMed: 27732330]
- 48.
- Sheu Y, Amati F, Schwartz AV, Danielson ME, Li X, Boudreau R, Cauley JA; Osteoporotic Fractures in Men (MrOS) Research Group. Vertebral bone marrow fat, bone mineral density and diabetes: The Osteoporotic Fractures in Men (MrOS) study. Bone. 2017 Feb 4;97:299-305. [PMC free article: PMC5367972] [PubMed: 28179169]
- 49.
- Greco EA, Lenzi A, Migliaccio S. Ther Adv Endocrinol Metab. 2015 Dec;6(6):273–86. The obesity of bone. [PMC free article: PMC4647134] [PubMed: 26623005]
- 50.
- Veldhuis-Vlug AG, Rosen CJ. Mechanisms of marrow adiposity and its implications for skeletal health. Metabolism. 2017;67:106–114. [PMC free article: PMC5325679] [PubMed: 28081773]
- 51.
- Ducy P, Zhang RR, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. [PubMed: 9182762]
- 52.
- Rosen CJ. Growth hormone, IGF-I and the elderly: Clues to potential therapeutic intervention. Endocrine. 1997 Aug;7(1):39–40. [PubMed: 9449029]
- 53.
- Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007 Jun;148(6):2635–2643. [PubMed: 17395698]
- 54.
- Poole KE, van Bezoolijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005 Nov;19(13):1842–1844. [PubMed: 16123173]
- 55.
- Appelman-Dijkstra NM, Papapoulos SE. Sclerostin inhibition in the management of osteoporosis. Calcif Tissue Int. 2016 Apr;98(4):370–380. [PMC free article: PMC4824823] [PubMed: 27016922]
- 56.
- Bandeira L, Lewiecki EM, Bilezikian JP. Expert Opin Biol Ther. 2017 Feb;17(2):255–263. Romosozumab for the treatment of osteoporosis. [PubMed: 28064540]
- 57.
- Albright F. Postmenopausal osteoporosis. JAMA. 1941;116:2465–2474.
- 58.
- Chestnut C, Notelovitz M, Clark G, Drinkwater B, Rosen C, Bell N, English S, Johnston CC, Cain D, Flessland K, Mallinak N. Use of the N-telopeptide of type I collagen to monitor the effect of therapy and predict changes in bone mineral density in postmenopausal women treated with hormone replacement therapy. Am J Med. 1997;102:29–37. [PubMed: 9209198]
- 59.
- Ettinger B, Pressman A, Sklarin P, Bauer D, Cualey JA, Cummings SR. Associations between low levels of serum estradiol, bone density and fractures among elderly women: SOF. J Clin Endocrinol Metab. 1998;83:2239–2243. [PubMed: 9661589]
- 60.
- Greendale GA, Edelstein S, Barrett-Connor E. Endogenous sex steroids and bone mineral density in older women and men. J Bone Miner Res. 1997;12:1833–1837. [PubMed: 9383688]
- 61.
- Smith EP, Boyd J, Frank GR, Takahasi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen receptor gene in a man. N Engl J Med. 1994;331:1056–1061. [PubMed: 8090165]
- 62.
- Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation in the physiological role of estrogens. J Clin Endocrinol Metab. 1995:3689–3698. [PubMed: 8530621]
- 63.
- Carani C, Qin K. simoni M, Faustini-Faustini S, Boyd J, Korach KS, Simpson ER. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. 1997;337:91–95. [PubMed: 9211678]
- 64.
- Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Lenrow DA, Holmes JH, Diewati A, Santana J, Rosen CJ, Strom BL. Effect of testosterone treatment on body composition and muscle strength in men over 65. J Clin Endo Metab. 1999;84:2647–2653. [PubMed: 10443654]
- 65.
- Dresner-Pollak R, Parker RA, Poku M, Thompson J, Seibel MJ, Greenspan SL. Biochemical markers of bone turnover reflect femoral bone loss in elderly women. Calcif Tissue Int. 1996;59:328–333. [PubMed: 8849397]
- 66.
- Food and Nutrition Board Institute of Medicine 2011; Dietary Reference Intakes for Calcium, Phosphorus, Magnesium Vitamin D and Fluoride. Washington DC, National Academy Press. www
.iom/vitaminD.edu. - 67.
- Storm D, Smith-Porter E, Musgrave KO, Vereault D, Patton C, Kessenich CR, Eslin R, Mohan S, Chen T, Holick MF, Rosen CJ. Calcium supplementation prevents seasonal bone loss and changes in biochemical markers of bone turnover in elderly New England women: A Randomized Placebo-Controlled Trial. J Clin Endocrinol Metab. 1998;83:3817–3826. [PubMed: 9814452]
- 68.
- Rosen CJ, Morrison A, Zhou H, Storm D, Hunter S, Musgrave KO, Chen T, et al. Elderly women in northern new England exhibit seasonal changes in bone mineral density and calciotropic hormones. Bone Miner. 1994;25:83–92. [PubMed: 8086854]
- 69.
- Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Mayne ST, Rosen CJ, Shapses SA. J Clin Endocrinol Metab. 2011 Jan;96(1):53–58. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. [PMC free article: PMC3046611] [PubMed: 21118827]
- 70.
- Kahwati LC, Weber RP, Pan H, Gourlay M, LeBlanc E, Coker-Schwimmer M, Viswanathan M. JAMA. 2018 Apr 17;319(15):1600–1612. Vitamin D, Calcium, or Combined Supplementation for the Primary Prevention of Fractures in Community-Dwelling Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. [PubMed: 29677308]
- 71.
- LeBoff MS, Kohlmeier L, Hurwitz S, Franklin J, Wright J, Glowacki J. Occult vitamin D deficiency in postmenopausal US women with acute hip fracture. JAMA. 1999;282:1505–1511. [PubMed: 10227320]
- 72.
- Rosen C, Donahue LR, Hunter S, Holick M, Kavookjian H, Kirshenbaum A, Mohan S, Baylink DJ. The 24/25kD serum insulin-like growth factor binding protein is increased in elderly women with fractures. J Clin Endocrinol Metab. 1992;74:24–28. [PubMed: 1370164]
- 73.
- Heaney RP, McCarron DA, Dawson Hughes B, Oparil S, Berga SL, Stern JS, Barr SI, Rosen CJ. Dietary Changes favorably affect bone remodeling in older adults. JADA. 99:1228–1233. [PubMed: 10524386]
- 74.
- Cushing H. Basophile adenomas of the pituitary body and their clinical manifestations. Bull Johns Hopkins Hop. 1932;50:137–145.
- 75.
- Thacker H. Glucocorticoid-induced Osteoporosis. Cleveland Clinic J Med 2010 77 843-844. [PubMed: 21186693]
- 76.
- Teegarden D, Proulx WR. martin BR. Peak bone mass in young women. J Bone Miner Res. 1995;10:711–715. [PubMed: 7639106]
- 77.
- Prior JC, Vigna Y, Schechter MT, Burgess AE. Spinal bone loss and ovulatory disturbances. N Engl J Med. 1990;323:1221–1227. [PubMed: 2215605]
- 78.
- Weaver CM, Gordon CM, Janz KF, Kalkwarf HJ, Lappe JM, Lewis R, O'Karma M, Wallace TC, Zemel BS. Osteoporosis Int. 2016 Apr;27(4):1281–386. The National Osteoporosis Foundation's position statement on peak bone mass development and lifestyle factors: a systematic review and implementation recommendations. [PMC free article: PMC4791473] [PubMed: 26856587]
- 79.
- Gilsanz V, Loro ML, Roe TF, Syrre J, Gilsanz R, Schulz EE. Gender differences in vertebral size in adults: biomechanical implications. J Clin Invest. 1995;95:2332–2337. [PMC free article: PMC295847] [PubMed: 7738196]
- 80.
- Bachrach LK, Guido D, Katzman D, Litt IF, Marcus R. Decreased bone density in adolescent girls with anorexia nervosa. Pediatrics. 1990;86:440–447. [PubMed: 2388792]
- 81.
- Johnston CC, Miller JZ, Slemenda CW, Reister TK, Hui S, Christian JC, Peacock M. Calcium supplementation and increases in bone mineral density in children. N Engl J Med. 1992;327:82–87. [PubMed: 1603140]
- 82.
- Menaa C, Vrtovsnik F, Freidlander G, Corvol M, Garbedian M. IGF-I a unique calcium dependent stimulator of 1,25 vitamin D production. J Biol Chem. 1995;270:25461–25467. [PubMed: 7592714]
- 83.
- Schacter GI, Leslie WD. Diabetes and Bone Disease. Endocrinol Metab Clin North Am. 2017 Mar;46(1):63–85. [PubMed: 28131137]
- 84.
- Shanbhogue VV, Mitchell DM, Rosen CJ, Bouxsein ML. Type 2 diabetes and the skeleton: new insights into sweet bones. Lancet Diabetes Endocrinol. 2016 Feb;4(2):159–73. [PubMed: 26365605]
- 85.
- McKay HA, Bailey DA, Wilkinson AA, Houston CS. Familial comparison of bone mineral density at the proximal femur and lumbar spine. Bone Miner. 1994;24:95–107. [PubMed: 8199536]
- 86.
- Smith DM, Nance WE, Kang KW, Christian JC, Johnston CC. Genetic factors in determining bone mass. J Clin Invest. 1973;52:2800–2808. [PMC free article: PMC302548] [PubMed: 4795916]
- 87.
- Seeman E. Genetic Determinants of the Population Variance in Bone Mineral Density. In The Aging Skeleton eds by CJ Rosen, J Glowacki and JP Bilezikian 1999; Academic Press San Diego, CA pp77-84.
- 88.
- Rogers J, Mahaney MC, Beamer WG, Donahue LR, Rosen CJ. Beyond one gene-one disease: Alternative strategies for deciphering genetic determinants of osteoporosis. Calcif Tiss Int. 1997;60:225–228. [PubMed: 9069155]
- 89.
- Utterlinden AG, Burger H, Huang Q, Fang Y. Relation of alleles of the collagen type I alpha 1 gene to bone density and the risk of osteoporotic fractures in postmenopausal women. N Engl J Med. 1998;338:1016–1021. MCGuigan FEA, Grant SFA, Hofman A et al. [PubMed: 9535665]
- 90.
- Hsu YH, Kiel DP. J Clin Endocrinol Metab. 2012 Oct;97(10) Clinical review: Genome-wide association studies of skeletal phenotypes: what we have learned and where we are headed. [PMC free article: PMC3674343] [PubMed: 22965941]
- 91.
- Richards JB, Zheng HF, Spector TD. Nat Rev Genet. 2012 Jul 18;13(8):576–88. Genetics of osteoporosis from genome-wide association studies: advances and challenges. [PubMed: 22805710]
- 92.
- Boyden LM, Mao J, Belsky J, et al. High bone density due to mutation in LDL-receptor related protein-5. New Engl J Med. 2002 May;346(20):1513–1521. [PubMed: 12015390]
- 93.
- Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, Peltonen L, Somer H, Hirose T, Dallapiccola B, De Pappe A, Swoboda W, Zabel B, Superti-Furga A, Steinmann B, Brunner HG, Jans A, Boles RG, Adkins W, van den Boogaard MJ, Olsen BR, Warman ML. Am J Hum Genet. 1996 Jul;59(1):146–51. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. [PMC free article: PMC1915094] [PubMed: 8659519]
- 94.
- Bonjour JP, Carrie AL, Ferrari S. Calcium enriched foods and bone mass growth in prepubertal girls: a randomized double blind placebo controlled trial. J Clin Invest. 1997;99:1287–1294. [PMC free article: PMC507944] [PubMed: 9077538]
- 95.
- Cadogan J, Blumensen A, Barker M, Eastell R. A longitudinal study of bone gain in pubertal girls: anthropometric and biochemical correlates. J Bone Min Res. 1998;13:1602–1612. [PubMed: 9783549]
- 96.
- Weaver CM, Peacock M, Johnston CC. Adolescent nutrition in the prevention of postmenopausal osteoporosis. J Clin Endocrinol Metab. 1999;84:1839–1843. [PubMed: 10372671]
- 97.
- Bonjour JP, Chevalley T. Endocr Rev. 2014 Oct;35(5):820–847. Pubertal timing, bone acquisition, and risk of fracture throughout life. [PubMed: 25153348]
- 98.
- Black DM, Rosen CJ. NEJM. 2016;374:257–262. Postmenopausal Osteoporosis.
- 99.
- Shoback D, Rosen CJ, Black DM, Cheung AM, Murad MH, Eastell R. J Clin Endocrinol Metab. 2020 Mar;105(3):587–594. Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society Guideline Update. [PubMed: 32068863]
- ABSTRACT
- INTRODUCTION
- EPIDEMIOLOGY OF OSTEOPOROSIS
- FACTORS THAT AFFECT BONE QUANTITY AND QUALITY
- BONE REMODELING AND ITS RELATIONSHIP TO BONE QUANTITY
- SYSTEMIC AND LOCAL ALTERATIONS IN THE BONE REMODELING SEQUENCE LEAD TO BONE LOSS
- PATHOGENIC FACTORS WHICH IMPAIR PEAK BONE MASS
- GENETIC DETERMINANTS OF PEAK BONE MASS
- SUMMARY
- REFERENCES
- Utilization of DXA Bone Mineral Densitometry in Ontario: An Evidence-Based Analysis.[Ont Health Technol Assess Ser....]Utilization of DXA Bone Mineral Densitometry in Ontario: An Evidence-Based Analysis.Medical Advisory Secretariat. Ont Health Technol Assess Ser. 2006; 6(20):1-180. Epub 2006 Nov 1.
- Review Osteoporosis: Clinical Evaluation.[Endotext. 2000]Review Osteoporosis: Clinical Evaluation.Lewiecki EM. Endotext. 2000
- Screening for the primary prevention of fragility fractures among adults aged 40 years and older in primary care: systematic reviews of the effects and acceptability of screening and treatment, and the accuracy of risk prediction tools.[Syst Rev. 2023]Screening for the primary prevention of fragility fractures among adults aged 40 years and older in primary care: systematic reviews of the effects and acceptability of screening and treatment, and the accuracy of risk prediction tools.Gates M, Pillay J, Nuspl M, Wingert A, Vandermeer B, Hartling L. Syst Rev. 2023 Mar 21; 12(1):51. Epub 2023 Mar 21.
- Review Unresolved issues in osteoporosis in men.[Rev Endocr Metab Disord. 2001]Review Unresolved issues in osteoporosis in men.Seeman E. Rev Endocr Metab Disord. 2001 Jan; 2(1):45-64.
- Circulating serum microRNAs including senescent miR-31-5p are associated with incident fragility fractures in older postmenopausal women with type 2 diabetes mellitus.[Bone. 2022]Circulating serum microRNAs including senescent miR-31-5p are associated with incident fragility fractures in older postmenopausal women with type 2 diabetes mellitus.Heilmeier U, Hackl M, Schroeder F, Torabi S, Kapoor P, Vierlinger K, Eiriksdottir G, Gudmundsson EF, Harris TB, Gudnason V, et al. Bone. 2022 May; 158:116308. Epub 2022 Jan 21.
- The Epidemiology and Pathogenesis of Osteoporosis - EndotextThe Epidemiology and Pathogenesis of Osteoporosis - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...