

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mapping Protein/DNA Interactions by Cross-Linking [Internet]. Paris: Institut national de la santé et de la recherche médicale; 2001.
Abstract
The highly conserved Ku heterodimer is implicated in double-strand break repair in many organisms. In addition, Ku-deficient yeast strains have pronounced telomere-specific phenotypes, including a significant shortening of the TG1-3 repeat on chromosome ends, disruption of subtelomeric silencing, and dispersion of the silencing factors from telomeric foci. By immunofluorescence and Chromatin ImmunoPrecipitation (ChrIP) studies, we show that the Ku heterodimer colocalizes with yeast telomeres. When DNA damage is induced by bleomycin, MMS, or a single double-strand cleavage (DSB), yKu80p and components of repressed subtelomeric chromatin (Sir proteins) are displaced from telomere clusters. The displacement of Sir proteins and yKu coincides with a slight drop in telomere proximal repression. ChrIP data confirm the immunofluorescence results and show that yKu is rapidly recruited to the sites of DSB, whereas Sir proteins are recruited with much slower kinetics. Sir recruitment to a DSB does not require yKu, Mre11, nor DNA ligase IV. Thus, probably none of the complexes specific for DSB repair, and probably not the repair event itself, is essential for Sir recruitment. The fact that Sir protein recruitment does not depend on the presence of yKu suggests that yKu--Sir interaction is modified when DNA damage is induced and may account for the release of Sir proteins from telomeres. The displacement of Sirs and yKu from telomeres, but not the recruitment of yKu to DSB, is curtailed in a rad9 and mec1 mutant. This suggests that the telomeric response is part of the checkpoint response mediated by the action of Mec1. The Mec1 protein itself associates with DSB in a yKu-independent manner but does not localize to telomeres, even in the presence of DNA damage, suggesting that Mec1p may act indirectly on telomeric proteins.
General Principle of Chromatin ImmunoPrecipitation (ChrIP)
ChrIP (chromatin immunoprecipitation) is a powerful tool for the spatial and temporal mapping of chromatin-bound factors in vivo. This technique allows one to determine whether and where in the genome a protein is bound, whether the interaction with DNA is direct or indirect (see Figure 1 for the general principle of the experiment). The initial step is the cross-linking of live cells with formaldehyde. Formaldehyde is a reagent that penetrates biological membranes readily, allowing the cross-linking to be done with intact cells, which reduces the risk of redistribution or reassociation of chromosomal proteins during the preparation of cellular or nuclear extracts. The chemical targets for formaldehyde are primary amino groups (lysine amino group and side chains of adenine, guanine, and cytosine), leading to the cross-linking of both protein--protein and protein--DNA. Both types of cross-links can be reversed by heating (65°C for protein--DNA, boiling for protein--protein).

Figure 1
General principle of Chromatin ImmunoPrecipitation assay.
After cross-linking, the cells are lysed, and crude extracts are sonicated to shear the DNA. Indeed, short DNA fragments provide higher mapping resolution and allow determination of the precise chromosomal location of chromatin-associated proteins. Extensive sonication is a way to generate fairly uniformly sized pieces of DNA. After preparation of the cellular extract and chromatin fragmentation, proteins together with cross-linked DNA are immunoprecipitated. Protein--DNA cross-links in the immunoprecipitated material are then reversed, and the DNA fragments are purified. If the protein under investigation is associated with a specific genomic region in vivo, DNA fragments of this region should be enriched in the immunoprecipitate (IP) compared to other portions of the genome. The presence of the relevant genomic regions in the IP is determined by PCR amplification with specific primers from the region in question and reference region. Comparison of the ratio of the PCR products from the region in question and reference region obtained in the IP relative to the input (IN = non-immunoprecipitated whole cell extract) allows quantification of the enrichment of the region of interest.
Important Steps to Control
DNA Shearing
This is a critical step because the extent of chromatin fragmentation will influence the resolution of the mapping experiment. To establish the conditions for sonication, a cellular lysate is prepared and subjected to various numbers of sonication cycles. Small aliquots are removed after each cycle, and finally DNA is isolated and analyzed by agarose gel electrophoresis and ethidium bromide staining to determine how many cycles of sonication are needed to shear to the desired size range. When choosing a certain size range for the DNA, one should also keep in mind that the amplification efficiency by PCR may decrease as the average fragment size approaches the distance between the PCR primers used.
Immunoprecipitation
The outcome of the experiment depends critically on the quality of the antibodies used (check for cross-reactions). The antibodies may need to be affinity purified for better specificity. The amount of antibody necessary for the quantitative immunoprecipitation of the protein of interest has to be determined. This can be done in pilot experiments in which increasing amounts of antibodies are combined with cell lysate. A control sample with no antibody should be included. The precipitated proteins and aliquots of the supernatant after IP are analyzed by Western blot to monitor in which condition the protein of interest is mainly found in the precipitate. It is necessary to control the efficiency of IP of each experiment by analyzing the protein content of the WCE (input), the supernatant, and the IP in Western blot. The salt and detergent composition of the buffers used for lysis, IP, and washing determines the stringency of the analysis. Increasing salt concentration or including 0.1% SDS will increase the stringency and reduce nonspecific IP. These conditions have to be adapted for each protein analyzed.
For each experiment, one might check the specificity of the ChIP by performing IP with no antibody or with a different antibody. This allows checking of the IP background due to nonspecific IP by the antibody support used to pull down (Sepharose, magnetic beads...). For DNA binding proteins, one should also perform a non-cross-linked control to check that no rebinding of the protein occurs during the precipitation steps.
PCR Analysis
To deduce whether a protein is associated with a particular genomic region, one compares the relative abundance of PCR products from the region in question relative to a reference. Therefore, it is important that the PCR reaction is quantitative.
Quantitative PCR should be simple. Theoretically, the amount of product doubles with each amplification cycle. However, this makes several false assumptions, the worst of which is that reaction occurs at 100% efficiency in all amplification cycles. In fact, inhibitors alter this efficiency, and later, amplification cycles of a typical PCR reaction have very low efficiencies compared to earlier cycles. At the start of a PCR reaction, reagents are in excess, template and product are at low enough concentrations that product renaturation does not compete with primer binding, and amplification proceeds at a constant, exponential rate. Exactly when the reaction rate ceases to be exponential and enters a linear phase of amplification is extremely variable, but it appears to be primarily attributable to product renaturation competing with primer binding (since adding more reagents or enzyme has little effect). At some later cycle, the amplification rate drops to near zero (plateau), and little more product is made (Figure 2). The consequence of this is that templates in a different initial amount can appear equally amplified if the last cycle of the PCR occurs at the plateau. Thus, it is necessary to collect quantitative data while the PCR is still in the exponential phase of amplification.
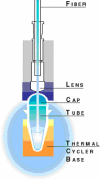
Figure 2
Scheme of the detection system.
1. Conventional PCR
The use of an internal control (IC) improves the reliability of the quantitative result, providing a means to monitor and correct for the efficiency of the PCR reaction. In this multiplex PCR, several PCR products from different primer pairs are amplified simultaneously in a single tube. The length of PCR product is generally <500 bp, with a size difference of approximately 50 bp between individual products. Thus, amplification efficiency should be biased minimally by size differences. One might check that the efficiency of amplification of the IC and product of interest (PI) are close. This could be done by quantifying the PCR products taken from a single sample at certain intervals during the amplification.
The PCR amplification might end in the exponential phase for both the internal control and the PCR PI. This can be tested by checking that the quantity of PCR products still increases in an exponential fashion along the last cycles. This assay is linear only over a very short range, and rare targets will possibly be below the limit of detection, whereas abundant targets will be past the exponential phase. To extend this range, duplicate reactions may be performed for a greater or lesser number of cycles or on serial dilutions of the sample.
Once these conditions are settled, then each reaction can be tested by checking that the PI/IC ratio is constant for PCR performed on serial dilution of the sample.
2. By Using Real-Time PCR
Quantitative real-time PCR is based on detection of a fluorescent signal produced proportionally during the amplification of a PCR product. This allows visualization directly of the exponential part of the PCR reaction.
2.1. System (see Figure 2)
In brief, the detection system consists of a thermal cycler connected to a laser and charge-coupled device (CCD) optics system. An optical fiber inserted through a lens is positioned over each well, and laser light is directed through the fiber to excite the fluorochrome in the PCR solution. Emissions are sent through the fiber to the CCD camera, where they are analyzed by the software's algorithms. Collected data are subsequently sent to the computer.
The software calculates the threshold cycle (Ct) for each reaction with which there is a linear relationship to the amount of starting DNA. Ct is the threshold cycle, i.e., the cycle number at which the reporter dye emission intensities rises above background noise (Figure 3) The Ct is determined at the most exponential phase of the reaction and is more reliable than end-point measurements of accumulated PCR products used by traditional PCR methods. The Ct is inversely proportional to the copy number of the target template; the higher the template concentration, the lower the threshold cycle measured.

Figure 3
One of the views available after completion of the run is an amplification window. This window shows the amount of fluorescence obtained in each amplification cycle for each reaction. The threshold cycle (Ct) is shown by the darker horizontal line.
There are many advantages to quantifying gene sequences using this technology, foremost being precision and sensitivity. This precision exists because the quantification of the gene sequence is determined by the Ct, which is calculated during the exponential phase of the reaction.
2.2. Real-Time Reporters: SYBR® Green, TaqMan®, and Molecular Beacons
All real-time PCR systems rely upon the detection and quantification of a fluorescent reporter, the signal of which increases in direct proportion to the amount of PCR product in a reaction.
SYBR® Green
In the simplest and most economical format, that reporter is the double-strand DNA-specific dye SYBR® Green (Molecular Probes). SYBR Green binds double-stranded DNA and, upon excitation, emits light. Thus, as a PCR product accumulates, fluorescence increases (Figure 4).

The advantages of SYBR Green are that it is inexpensive, easy to use, and sensitive. The disadvantage is that SYBR Green will bind to any double-stranded DNA in the reaction, including primer dimers and other nonspecific reaction products, which results in an overestimation of the target concentration. For single PCR product reactions with well-designed primers, SYBR Green can work extremely well, with spurious nonspecific background showing up only in very late cycles.
TaqMan® and Molecular Beacons
Both are hybridization probes relying on fluorescence resonance energy transfer (FRET) for quantification. The probe is designed to anneal to the target sequence between the traditional forward and reverse primers (Figure 5).

TaqMan Probes are oligonucleotides that contain a reporter fluorochrome [usually 6-carboxyfluorescein (6-FAM)] and a quencher fluorochrome [6-carboxy-tetramethyl-rhodamine (TAMRA)] added at any T position or at the 3' end. When irradiated, the excited fluorochrome transfers energy to the nearby quenching dye molecule rather than fluorescing, resulting in a nonfluorescent substrate. TaqMan probes are designed to hybridize to an internal region of a PCR product and to have a higher Tm than the primers, but during the extension phase, the probe must be 100% hybridized for success of the assay. During PCR, when the polymerase replicates a template on which a TaqMan probe is bound, the 5' exonuclease activity of the polymerase cleaves the probe. This separates the fluorescent and quenching dyes, and FRET no longer occurs. The amount of fluorescence released during the amplification cycle is proportional to the amount of product generated in each cycle.
Molecular beacons also contain fluorescent and quenching dyes, but FRET only occurs when the quenching dye is directly adjacent to the fluorescent dye. Molecular beacons are designed to adopt a hairpin structure while free in solution, bringing the fluorescent dye and quencher in close proximity. When a molecular beacon hybridizes to a target, the fluorescent dye and quencher are separated, FRET does not occur, and the fluorescent dye emits light upon irradiation. Unlike TaqMan probes, molecular beacons are designed to remain intact during the amplification reaction and must rebind to target in every cycle for signal measurement.
TaqMan probes and molecular beacons allow multiple DNA species to be measured in the same sample (multiplex PCR), because fluorescent dyes with different emission spectra may be attached to the different probes. Multiplex PCR allows internal controls to be co-amplified. These hybridization probes afford a level of discrimination impossible to obtain with SYBR Green, because they will only hybridize to true targets in a PCR and not to primer dimers or other spurious products.
Investing in the Real-Time Technique
Real-time PCR requires an instrumentation platform that consists of a thermal cycler, computer, optics for fluorescence excitation and emission collection, and data acquisition and analysis software. These machines, available from several manufacturers, differ in sample capacity (some are 96-well standard format, others process fewer samples or require specialized glass capillary tubes), method of excitation (some use lasers, others use broad spectrum light sources with tunable filters), and overall sensitivity. There are also platform-specific differences in how the software processes data.
SYBR is a registered trademark of Molecular Probes. TaqMan is a registered trademark of Roche Molecular Systems. ABI PRISM is a registered trademark of PE Applied Biosystems.
Appendix: Chromatin-IP (ChrIP) Protocol
Formaldehyde Cross-linking
- add 1.5 ml of formaldehyde 36% (stock solution) to 50 ml of culture (= 1% final)
- shake slowly at 30°C for 15 min
- add 2.5 ml of glycine 2.5 M
- shake for 5 min
- spin down
- wash 1X with PBS 1X
- freeze pellet quickly in liquid nitrogen
Crude Extract
- add 0.5 g glass beads
- add 400 μl of lysis buffer
- bead beater 3X for 20 sec (check for lysis under microscope)
- put tube with hole at bottom on top of 3-ml tube in 13-ml tube and spin to recover supernatant
- sonicate 3X for 20 sec (settings: 4, hold, constant, but output = about 20 => 500 bp-1.5 kb DNA fragments)
- transfer to Eppendorf tube
- spin at 7,000 rpm for 2 min in a cold room
- recover supernatant
- spin at 7,000 rpm for 2 min in a cold room
- recover supernatant
WCE
- save 25-μl aliquots as INPUT, freeze at --20°C until cross-linking reversal step
INPUTs
- save 5-μl aliquots for Western
- input aliquots for Western
IP
- wash 40 μl per sample Dynabeads with PBS 1X, 5 mg/ml BSA shaking for 30 min
- add 5 μl of 9E10 antibody per sample or elution of strips for affinity-purified sera
- incubate 1 hour to overnight at 4°C
- wash bound beads twice with PBS 1X, 5 mg/ml BSA shaking for 5 min at 4°C
- resuspend beads in 40 μl/sample PBS 1X, 5 mg/ml BSA
- add 40 μl of beads to each sample
- incubate 1 hour to overnight at 4°C
- remove beads with magnetic device
- wash twice with 600 μl of lysis buffer, shaking 5 min at 4°C
- wash 1X with 600 μl of wash buffer, shaking 5 min at 4°C
- wash 1X with 600 μl of TE, shaking 1 min at 4°C
- resuspend beads in 60 μl of TE/1% SDS
- incubate 10 min at 65°C with shaking
- spin 5 sec at 13,000 rpm, put on magnet device
- save supernatant in a new Eppendorf tube
- save 5-μl sample for Western
IPs, Aliquots for Western
- resuspend beads again in 60 μl of TE/1% SDS
- incubate 10 min at 65°C with shaking
- spin 5 sec at 13,000 rpm, put on magnet device
- save supernatant in same Eppendorf tube as before
Cross-Linking Reversal and Recovery of DNA
- add 130 μl of TE/1% SDS to IPs/100 μl to INPUTs
- submerge at 65°C for 6 hours to overnight
- add 240 μl of TE and 20 μl of proteinase K (10 mg/ml) to IPs
- add 120 μl of TE and 10 μl of proteinase K (10 mg/ml) to INPUTs
- incubate 2 hours at 37°C
- add 250 μl of TE to INPUTs
- add 50 μl of LiCl 5M
- phenol extraction 1X
- add 2 μl of glycogen (20 mg/ml)
- ethanol precipitation
- resuspend DNA in 60 μl of TE/INPUTs in 30 μl of TE/IPs
Solutions
2.5 M glycine: 18.8 g of glycine in 100 ml of H2O
Lysis Buffer
- 5 ml of HEPES/KOH 1 M, pH 7.5, 50 mM
- 2.8 ml of NaCl 5 M, 140 mM
- 0.2 ml of sodium-EDTA 0.5 M, 1 mM
- 1 ml of 100% Triton X-100, 1%
- 0.1 g of sodium deoxycholate, 0.1%
- H2O to 100 ml
- add protease inhibitors
LiCl 5 M
- 21.2 g of LiCl
- 5 ml of 1 M Tris, pH 8.0
- H2O to 100 ml
Wash Buffer (Store at Room Temperature)
- 1 ml of 1 M Tris, pH 8.0
- 5 ml of 5 M LiCl
- 500 μl of 100% NP-40
- 0.5 g of sodium deoxycholate
- 200 μl of 0.5 M sodium-EDTA
- H2O to 100 ml
- add protease inhibitors
Websites:
http://www.appliedbiosystems.com/molecularbiology/about/pcr/sds/
References
- Hecht A, Grunstein M. Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol. 1999;304:399–414. [PubMed: 10372373]
- Dedon P C, Soults J A, Allis C D, Gorovsky M A. A simplified formaldehyde fixation and immunoprecipitation technique for studying protein--DNA interactions. Anal Biochem. 1991;197:83–90. [PubMed: 1952079]
- Heud C A, Stevens J, Livak K J, Williams P M. Real time PCR. Genome Res. 1996;6:986–94. [PubMed: 8908518]
- Gibson U E M, Heid C A, Williams P M. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. [PubMed: 8908519]
- PubMedLinks to PubMed
- Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast.[Cell. 1999]Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Cell. 1999 May 28; 97(5):621-33.
- Ku Binding on Telomeres Occurs at Sites Distal from the Physical Chromosome Ends.[PLoS Genet. 2016]Ku Binding on Telomeres Occurs at Sites Distal from the Physical Chromosome Ends.Larcher MV, Pasquier E, MacDonald RS, Wellinger RJ. PLoS Genet. 2016 Dec; 12(12):e1006479. Epub 2016 Dec 8.
- Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing.[EMBO J. 1998]Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing.Boulton SJ, Jackson SP. EMBO J. 1998 Mar 16; 17(6):1819-28.
- Review The Ku heterodimer: function in DNA repair and beyond.[Mutat Res Rev Mutat Res. 2015]Review The Ku heterodimer: function in DNA repair and beyond.Fell VL, Schild-Poulter C. Mutat Res Rev Mutat Res. 2015 Jan-Mar; 763:15-29. Epub 2014 Jul 4.
- Review Telomere biology: integrating chromosomal end protection with DNA damage response.[Chromosoma. 2005]Review Telomere biology: integrating chromosomal end protection with DNA damage response.Slijepcevic P, Al-Wahiby S. Chromosoma. 2005 Sep; 114(4):275-85. Epub 2005 Oct 15.
- Examining the Distribution of Telomeric and DNA Repair Proteins by ChrIP and Rea...Examining the Distribution of Telomeric and DNA Repair Proteins by ChrIP and Real-Time PCR - Mapping Protein/DNA Interactions by Cross-Linking
Your browsing activity is empty.
Activity recording is turned off.
See more...