

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Arvin A, Campadelli-Fiume G, Mocarski E, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007.

Introduction
Foreign material entering multicellular organisms triggers a range of defense reactions which, when successful, subjugates and removes the invaders. Invertebrates and plants have natural defense systems, which recognize commonly shared patterns and usually react in a stereotypical manner. Long-lived animals such as vertebrates add to these natural defenses with adaptive systems that show discriminating recognition machinery, complex and varying effector mechanisms and development of persistent or “memory” responses. Under ideal circumstances, immune defense proceeds with minimal or inapparent damage to the host. In other situations, the defense system is less successful and the host tissues become damaged by the reaction. We usually consider the former situation as immunity and the latter as immunopathology. However, in both instances, mechanisms at play may be similar and deciding if the process is one or the other may require Lucretian logic.“What is food to one man is bitter poison to others” Lucretius De Rerum Natura
(50BCE)
With microorganisms, the commonest circumstance that results in immunopathology is where the microbe persists and continues to cause an innate and adaptive response. These, however, prove ineffective to remove or neutralize the agent. Thus the reaction becomes chronic and host tissues become damaged as a consequence. This situation occurs in tuberculosis as well as hepatitis B and C virus infections. Over time, many microbes with a long association with a host species find ways of persisting by evading responses that would either eliminate them or cause too much tissue damage. Human CMV infection in immunocompetent adults provides an example of this scenario (Reddehase, 2002). Other circumstances that result in immunopathology involve settings where one or more components of normal immune defense are compromised for genetic or other reasons. Prolonged severe genital herpes simplex virus (HSV) lesions in AIDS patients with very low CD4+ T-cells represent such an example (Koelle and Corey, 2003). Atopics too often have problems clearing HSV and hence often develop skin and eye lesions (Pepose, 1991a). In addition, some microbes are considered as able to trigger immune reactions that target host components themselves (autoimmune disease) or cause infected cells to undergo neoplastic transformation. The herpesvirus EBV provides an example of the latter in genetically susceptible individuals (Kieff and Rickinson, 2001). There are no undisputed examples wherein herpesviruses cause autoimmune diseases. However, HHV-6 has been proposed to cause multiple sclerosis (Swanborg et al., 2003) and HSV infection may cause an autoimmune corneal inflammatory lesion (Streilein et al., 1997).
Herpes simplex virus is a pathogen that only rarely appears to be involved in immune mediated tissue damage. Characteristically, primary or recurrent infections at superficial mucosal or dermal sites result in viral replication and destruction of most cells that support infection. This process induces an innate inflammatory reaction that contributes to infection control. Some cells, likely Langerhans dendritic cells, leave the site and carry viral antigens to draining lymph nodes where an adaptive response is induced or recalled. After a few days, effectors of adaptive immunity are recruited to the site, initially CD4+ T-cells followed by CD8+ cells, and these T-cells, probably assisted by antibody, complete the task of recovery (Koelle and Corey, 2003). Virus is removed and the inflammatory reaction subsides usually without trace. These events can be judged to represent immunity. When T cell function is impaired, as can happen in AIDS patients, HSV removal is impaired and the inflammatory reaction becomes unusually severe and prolonged. This situation can be taken to represent immunopathology.
Certain tissue sites are particularly vulnerable to damage by an inflammatory response. These are sites where virus is difficult to dislodge, so the inflammatory reaction becomes prolonged and destructive, or where tissue repair leaves a functionally damaged organ. The eye is the site which best exemplifies such circumstances. In this organ, where inflammation or scar tissue along the visual axis impairs function, HSV infection may permanently impair vision. The most frequent example is herpetic stromal keratitis (SK). This chronic inflammatory reaction damages the stroma and can become sufficiently severe to merit corneal transplantation. About 20% of ocular HSV infections in humans result in stromal keratitis (Pepose et al., 1996). In most instances, these are caused by HSV-1 and result from reactivation from latent infection in the trigeminal ganglion (Pepose et al., 1996). Most of our understanding of the immunopathogenesis of SK comes from studies in animal models, in former times mostly the rabbit and now most usually the mouse.
Human HSV infections may also cause anterior as well as posterior uveitis (retinitis). These are rarer manifestations of HSV infection and much of the damage may result from direct viral damage more than from immunopathology. However, uveitis lesions in the rabbit and mouse models have a definite immunopathological component. Finally, HSV is the cause of a vision destroying reaction in the retina termed acute retinal necrosis (ARN). This lesion occurs both in children infected neonatally or later with HSV-2 as well as in adults where HSV-1 is usually involved (Margolis and Atherton, 1996). Acute retinal necrosis has also been studied in animal models, where lesions were shown to be immunopathological in part (Atherton, 2001).
Inflammatory reactions associated with HSV infection in the peripheral and central nervous systems may also be judged as immune mediated. The best studied example is ganglionitis, an HSV induced lesion that occurs in heterologous hosts but may not be a feature of the natural human disease. Lesions caused by HSV in the CNS are the most dramatic and devastating manifestations of HSV infection of humans. This rare disease is usually caused by HSV-1 infection in adults and is mostly a direct virologic lesion. However, immunopathological events such as demyelination may also occur in a few cases. In infants and children, encephalitis is more commonly associated with neonatal infection with HSV-2 and this lesion appears to be the direct result of a lytic virus infection.
Finally, there are some chronic inflammatory reactions that have been associated with HSV infection especially with the widespread use of modern technology to detect viral DNA. Some reports suggest that the virus or an immune response against it accounts for such HSV associated diseases as erythema multiforme, arteritis, Alzheimer’s disease, Bell’s palsy and Behcet’s disease.
Herpes infections and ocular disease
At least four human herpesviruses have been implicated as causes of ocular disease. Two alphaherpesviruses, HSV and varicella zoster virus (VZV), the betaherpes virus CMV and the gammaherpesvirus EBV. CMV is a cause of retinitis, a lesion found only in immunosuppressed individuals, the majority of which were formerly AIDS patients, but now transplant recipients, especially recipients of bone marrow (Holland et al., 1996). With the widespread use of protease inhibitors to control HIV, CMV retinitis is now mainly a disease of transplant recipients. The lesion itself is likely a direct consequence of viral replication in retinal cells. EBV is an occasional cause of SK lesions. These are characterized by an abundance of lymphoma like cells, in the stroma that are presumed to be mainly B cells (Matoba, 1990).
More commonly both HSV and VZV cause lesions in the anterior segment, principally the cornea. Both HSV and VZV can also cause uveitis and acute retinal necrosis (ARN).
Keratitis in humans
Both HSV and VZV can infect multiple structures in the eye. Lesions caused by HSV are much more common. The incidence of HSV ocular disease ranges from 4.1–20.7 cases/100 000 patient years representing the commonest single infectious cause of vision impairment in the western world (Pepose et al., 1996). Of the three general types of HSV corneal disease, Infectious Epithelial Keratitis (IEK) is the most common lesion and appears to be a result of the direct effect of viral infection. Both disciform keratitis (HSV endotheliitis) and SK are thought to be mainly the consequence of immune mediated mechanisms rather than direct viral damage. IEK lesions are a result of viral replication and spread in the superficial epithelial layer of the cornea. This condition is usually self-limiting and no permanent corneal damage results. The quick remission seen with timely antiviral therapy suggests a simple viral cytolytic mechanism. However, virus invariably infects nervous ramifications in the cornea that have free ends within the epithelial layer, thus allowing retrograde transport and establishment of latency (Shimeld et al., 2001). In addition, as a consequence of epithelial damage, virus can spread to the underlying stromal keratocytes and cause what is usually termed a necrotizing form of stromal keratitis (Liesegang, 1999). This terminology is not used by many ophthalmologists since necrosis also occurs in immune mediated SK (T. P. Margolis, personal communication, 2003).
Disciform keratitis (DK) is a lesion in which the corneal endothelium is the primary site of damage. This form of ocular disease appears immunopathological based upon the fact that early intervention with corticosteroids leads to complete resolution (Liesegang, 1999). In DK, the inflammatory reaction of the endothelium sometimes results in secondary stromal and epithelial edema but there is usually no stromal infiltrate or neovascularization. One of the characteristic findings is the demonstration of keratic precipitates or KP (Liesegang, 1999). The exact nature of the KP is unknown but they could be aggregates of macrophages or NK cells attracted by the immunoglobulins on the surfaces of infected cells (Liesegang, 1999). An alternative idea is that KP represent cytotoxic T-cells recognizing viral epitopes on the endothelial cells (Liesegang, 1999). The role of live virus in disease development is supported by finding antigens, live virus and DNA in the anterior chamber and perhaps also corneal endothelial cells (Kaufman et al., 1971; Sundmacher and Neumann-Haefelin, 1979a). It has been postulated that productive infection of the endothelial cell elicits a cellular and humoral immune response (Sundmacher and Neumann-Haefelin, 1979b), but this evidence is only circumstantial. Alternative suggestions include a possible delayed type hypersensitivity reaction to persisting HSV antigens within the stroma or the endothelium (Pepose, 1991b). It is difficult to resolve the nature of DK pathogenesis since animal models to study it are less than ideal. Disciform disease is seen in rabbits with an intracorneal injection of soluble viral antigen (Williams et al., 1965). Using the rabbit model for DK, some have suggested that the lesions involve immune complex formation and antibody dependent cell mediated cytotoxicity (Meyers and Chitjian, 1976).
Inflammation of the corneal stroma (SK) as a result of HSV-1 (rarely HSV-2) infection can lead to a blinding immuno-inflammatory lesion of the stroma. This only accounts for approximately 2% of initial episodes of ocular disease but approximately 50% of recurrent ocular HSV disease (Norn, 1970). A similar, but even more devastating lesion can be caused by VZV infection. Fortunately, this is quite rare and also usually occurs as a consequence of reactivation (zoster). The infections usually heal quickly unless the patient is immuno-suppressed (Pepose et al., 2003). Recurrent lesions can be very severe and most difficult to treat and control. Frequently, corneal lesions are accompanied by conjunctivitis, anterior uveitis and lipid keratopathy (Pepose et al., 2003). If the virus is not controlled, it spreads to involve the iris and the corneal stroma. Stromal lesions can become sclerotic and very persistent and is believed to be immune mediated, however, the mechanism is not known and is difficult to study. Patients often lose sensitivity of the cornea and involuntary physical damage can result in secondary bacterial infection.
Several observations suggest the operation of an immune etiology behind HSV induced SK (see Table 35.1). These include the fact that the lesions are persistent and are manifest well beyond the time that virus or viral antigens can be demonstrated. Lesions often need to be managed with indefinite corticosteroid treatment and reactivation lesions, except initially, do not benefit from acyclovir antiviral treatment. Also making a case for the pathogenesis of SK involving immunopathology is the fact that the lesion is very uncommon in immunosuppressed patients. Finally, clones of T-lymphocytes reactive to viral epitopes and possessing cytotoxic activity can be cultured from corneas showing chronic SK lesions (Verjans et al., 1998; Koelle et al., 2000).
Table 35.1
Immunopathological basis for SK in humans.
Approximately 90% of patients maintain good visual acuity despite prolonged disease. However, in many cases resolution of inflammation is associated with a permanent loss of vision resulting from corneal scarring and ulceration. This necessitates treatment by corneal transplantation, which in itself can sometimes be a high risk factor for recrudescent herpetic keratitis (also called newly acquired herpetic keratitis) (Remeijer et al., 1997) and super-infection with a different strain (Remeijer et al., 2002)
The corneal stroma may be affected by several mechanisms; this may be secondary to disease of the epithelium (IEK) or endothelium (DK) or as a stromal edema resulting from a damaged endothelium. In humans, SK manifests itself in two primary forms that are perhaps mis-termed necrotizing SK and immune SK (Liesegang, 1999). While the former is thought to result from direct viral invasion of the stroma, chronic immune mechanisms, possibly of an autoimmune nature (yet unproven), are suspected in pathogenesis of the latter (Pepose et al., 1996). These divisions are not mutually exclusive and necrosis can definitely occur in the immune form of disease. Intact virions and antigens can be detected in corneal keratocytes, endothelial cells and foci of epithelial cells in specimens from patients with acute (necrotizing) stromal keratitis (Kobayashi et al., 1972; Metcalf and Kaufman, 1976). This suggests that replicating virus and the resulting host inflammatory response leads to stromal cell destruction. This acute necrotizing form of SK eventually may become chronic, then considered to be the immune form of SK, when viral antigens are no longer present. The signs of SK are generally quite variable but they include the influx of a large number of different kinds of cells including polymorphonuclear leukocytes (PMN), macrophages, Langerhans’ cells, natural killer (NK) cells, plasma cells and T-lymphocytes (Pepose et al., 1985a; Youinou et al., 1985, 1986; Miller et al., 1993). In chronic herpetic SK in humans, the predominant population are macrophages and T-lymphocytes (Youinou et al., 1985). Excess neovascularization also occurs in some patients.
The original mechanism proposed for the pathogenesis of the immune form of SK focused on the role of anti-HSV antibodies. This was based on the finding that rings (Wessely rings) seen in the mid-stroma of the cornea in immune stromal keratitis were positive for IgM, IgG and IgA (Meyers-Elliot et al., 1980). Herpes virus particles have been demonstrated in these rings, many of them defective or incomplete (Meyers-Elliot et al., 1980). In addition, viral antigens have been found localized in the keratocytes of the corneal stroma in transplanted corneas (Youinou et al., 1986; Easty et al., 1987). Hence it has been speculated that viral antigens trapped in the stroma acted as a nidus for deposition of anti-HSV antibodies that fix complement and leads to cellular damage (Pepose et al., 1996). Viral antigens can also be presented to the infiltrating T-lymphocytes. In clinical specimens, increased levels of class Ⅰ and Ⅱ HLA antigens have been noted in areas of the greatest infiltrate, suggesting active presentation of antigens (Pepose et al., 1985b). Both CD4+ and CD8+ T-cells occur in chronic herpetic SK with the former dominating the total T-cell numbers (Youinou et al., 1986). Most of these cells are reactive against HSV antigens with the CD4+ subset reactive to peptide epitopes from UL21 and UL49 tegument proteins of HSV (Verjans et al., 1998; Koelle et al., 2000). They do not apparently recognize antigens derived from corneal tissues which would provide evidence for an auto-immune mechanism (Verjans et al., 1998). Corneal derived CD4+ cells have been shown to possess cytotoxic activity suggesting the possible operation of this mechanism in stromal cell injury (Verjans et al., 1998; Koelle et al., 2000).
Animal models for SK
Understanding the pathogenesis of human SK from clinical observations, transplant material and the occasional samples obtained at biopsy is difficult. Fortunately, convenient animal models exist wherein HSV infection of the eye reproducibly generates a stromal inflammatory response. Moreover, this appears to reflect human immune SK at least before it is treated. The usual animal models are the mouse and rabbit with the latter now rarely used except for studies on therapy. Events in SK pathogenesis are mainly studied in primary infection of various mouse strains. Since human SK is most commonly a sequel to reactivated HSV, a better animal model should be one where lesions follow reactivation. Such models have in fact, been described for both mice and rabbit (Myers-Elliot and Chitjian, 1983; Shimeld et al., 1989) but these are expensive and inconvenient and have contributed minimally to the understanding of pathogenesis. Rabbit reactivation can be achieved by ocular iontophoresis but this seldom gives rise to SK lesions (Myers-Elliot and Chitjian, 1983). The mouse reactivation model can be achieved by infecting mice under a cover of neutralizing antibody, then after some weeks asymptomatic animals are exposed to UV light. In usually a minority of animals, virus reactivates and generates an inflammatory reaction in the stroma (Shimeld et al., 1989). Few papers have employed the model, and the results of these usually support the basic findings of the primary infection model; namely that SK is an immuno-inflammatory lesion mainly orchestrated by CD4+ T cells (Shimeld et al., 1996).
The primary infection model usually uses strain HSV–1 RE and involves virus application to a lightly scratched cornea. Replication begins in epithelial cells of the cornea and usually the conjunctiva, but in immuno-competent mice rarely spreads to involve stromal cells or cells in uveal tissue. Characteristically, the viral replication events are over by 5–6 days and viral gene expression, as judged by protein detection or viral mRNA, are undetectable beyond a further 2–3 days (Babu et al., 1996). Viral DNA, however, can be detected for prolonged periods although copy numbers, detectable by real-time PCR, do not exceed 2000–5000 copies per cornea by 14 days p.i. (our unpublished results). When looked at with an ophthalmoscope, the initial viral replication events are accompanied by a barely detectable inflammatory reaction with new blood vessel growth from the limbus (the location of blood vessels at the edge of the vessel free normal cornea) the most obvious feature. This is often referred to as the preclinical phase, although in fact with appropriate tests is readily observable.
Innate reactions to infection in the mouse model
HSV infection of the corneal epithelium sets off a range of humoral and cellular events that taken together help contain infection. Unfortunately, some of these also set the stage for subsequent immunopathology. A prominent early cellular event is the influx of polymorphonuclear neutrophils (PMN). This occurs mainly into stromal tissues subjacent to the infected epithelium. Such PMN escape from blood vessels at the limbus presumably in response to signaling molecules generated from virus infected cells. The nature of such signaling molecules is unclear but several chemokines, including those known to be chemotactic to PMN, can be demonstrated within 12 hr p.i. (Su et al., 1996; Thomas et al., 1998)
The PMN response is at its peak around 48 hrs and it seems that this response helps control viral replication. Thus depleting PMN with specific monoclonal antibodies results in more intense and prolonged virus infection in the cornea (Tumpey et al., 1996; Thomas et al., 1997). Moreover, PMN suppressed animals may succumb to encephalitis since virus now spreads to the brain. Such observations indicate that PMN are part of the antiviral defense system although it is unclear how this function is performed. Accordingly, virus infected cells and PMN are usually not in direct contact implying that the protective function is indirect. Ideas for the mediation of such defenses include IFNγ and TNFα production as well as nitric oxide production by PMN (Daheshia et al., 1998a). This topic has not been fully explored using, for example, knockout mice and other means of implicating potential antiviral mechanisms.
The PMN response to virus is not only a defense reaction. Indeed products released from PMN have been proposed to contribute to corneal damage possibly unmasking autoantigens subsequently involved in the immunopathology (Thomas et al., 1997). In addition, PMN contribute to the process of neovascularization, a prominent feature of SK and a necessary step in its pathogenesis (Zheng et al., 2001a; Lee et al., 2002a). It appears that PMN may be a source of angiogenesis factors such as VEGF as well as tissue degrading enzymes which breakdown the stromal matrix and facilitates the growth of new blood vessels. One such enzyme released by the granules of activated PMN is MMP9 (Lee et al., 2002a). Since infected mice given the MMP-9 inhibitor TIMP-1 as well as MMP9-/- mice have reduced angiogenic and SK responses, MMP9 appears to be intricately involved in SK pathogenesis (Lee et al., 2002a).
Although PMN dominate the early inflammatory reaction to ocular HSV infection, other cell types can also be demonstrated. These include macrophages, dendritic cells (DC), NK cells but not B or αβ TCR T cells. The roles for these other cell types have received minimal investigation. It is likely, however that the macrophage is a source of angiogenic factors such as VEGF and FGF as well as the angiogenic CXC chemokines. For example MIP-2 (CXCL8) appears important since infected mice lacking the receptor for MIP-2 have an impaired PMN response (Banerjee et al., 2004a). In addition, in vivo neutralization of MIP-2 in HSV infected mice reduces PMN migration (Yan et al., 1998). Macrophages, along with DC, also act as a source of cytokines demonstrable early after infection. Most prominent of these are IL-1 and IL-6, both of which can also be produced by virus infected epithelial cells themselves (Tran et al., 1998; Kanangat et al., 1996). Indeed, it could be that these two cytokines are critical signaling molecules responsible for the many paracrine events set off by virus infected epithelial cells. The other early events described include the production of IL-12, VEGF and TNFα, but none of these are thought to be products of virus-infected cells themselves (Zheng et al., 2001b; Kumaraguru and Rouse, 2002). We have demonstrated that IL-6, for example, can cause macrophages in vitro to generate VEGF (Banerjee et al., 2004a), and IL-1 is well known to cause mononuclear cells to produce TNFα and other cytokines (Neta et al., 1992). Our recent findings also indicate that within HSV infected corneas, IL-1 maybe responsible for IL-6 expression, which in turn upregulates VEGF production (Biswas et al., 2004).
The cytokine IL-12 appears as a pivotal molecule in SK pathogenesis. Knockout mice, for example, unable to produce IL-12 have only mild SK lesions (Osorio et al., 2002). The source of IL-12 following HSV infection remains to be clarified, since as mentioned it does not appear to be HSV-infected cells themselves (Kumaraguru and Rouse, 2002) However conceivably viral DNA that has pathogen associated molecular pattern (PAMP) activity could represent such a stimulus (Zheng et al., 2002). The most likely producer cell types are DC and macrophages. The DC initially involved would seem to be the resident cells only recently demonstrated as present in normal non-inflamed corneas (Hamrah et al., 2003). A prominent feature of the injured cornea, including that caused by HSV, is the invasion of Langerhans DC, likely from the conjunctiva, into the cornea (Jager et al., 1991). However, this event takes several days to occur. Likely such cells also act as a source of cytokines and chemokines but their major function in SK pathogenesis is transport of viral antigens to lymphoid tissue where the adaptive immune response is initiated (discussed later).
The cytokine IL-12 has several downstream effects that impact on SK pathogenesis. The primary effect is induction of IFNγ production by cells with IL-12 receptors. Although not proven in the eye, the most likely cells that respond and produce IFNγ are natural killer (NK) cells. Such cells in non-ocular systems have been shown to be important for resistance to HSV. In fact, removing them results in heightened susceptibility (Rager-Zisman et al., 1987). An early study of SK indicated that NK removal ameliorated SK (Tamesis et al., 1994; Bouley et al., 1996), although this issue warrants further investigation. Whatever the source of IFNγ, this molecule appears to be intricately involved in antigen processing as well as other events critical for SK pathogenesis. These include up-regulation of the cell adhesion molecule PECAM-1 on vascular endothelial cells, at the limbus (Tang and Hendricks, 1996). This is a necessary step for normal PMN invasion as evidenced by the fact that neutralization of IFNγ or PECAM-1 results in diminished PMN ingress (Tang and Hendricks, 1996).
The importance of IFNγ in facilitating cell migration is further underscored by studies with human corneas. Stimulation of human corneal cells in vitro with IFNγ, and also IL-1 and TNFα, rapidly up-regulates ICAM-1 expression (another cell adhesion molecule that participates in the adhesion and extravasation of cells) (Pavilack et al., 1992). IFNγ also upregulates MHC Class Ⅱ expression on the antigen presenting cells involved in the induction of the initial antigen specific CD4+ T-cell response in local draining lymph nodes (Dreizen et al., 1988; Foets et al., 1991). On the other hand, IFNγ could help modulate lesion development since it also induces angiostatic chemokines such as IP-10 and MIG (Lee et al., 2002b). Accordingly the IL-12 response to HSV infection indirectly impacts on both inflammatory and regulatory effects on SK.
In Fig. 35.1 several critical events are shown that are set into play by HSV during the first 6–7 days postinfection. By the end of this often-called preclinical phase, the corneal tissues show little or no damage. The epithelium is fully intact, the stroma has few if any inflammatory cells and cytokine/chemokine levels have fallen significantly. The most obvious sign of change is a neovascular bed that continues to expand slowly beyond the limbal region. Nevertheless, in spite of the quiet appearance, notable changes begin to occur which constitute the true immunopathological events of SK. Accordingly, the T-cell orchestrators begin to invade via the new blood vessels and an intense inflammatory response ensues. This becomes obvious upon ophthalmoscopic examination and is frequently referred to as the clinical phase.

Fig. 35.1
Some early critical events occurring after HSV-1ocular infection.
Adaptive immunity in SK
Migration of T-cells that express appropriate homing molecules escaping from the newly established blood vessels represents a crucial step in SK pathogenesis. Mice without T-cells never develop typical SK lesions (Metcalf et al., 1979; Mercadal et al., 1993), but do if given T-cell transfers (Russell et al., 1984; Mercadal et al., 1993). Although debated early on, most investigators now agree that CD4+ T-cells, with the type 1 producing cytokine phenotype, are the main aggressors in HSK (Niemialtowski and Rouse, 1992; Mercadal et al., 1993). Such cells trigger the invasion of non-specific inflammatory cells, surprisingly once again dominated by PMN, giving rise to a peak response around 15 days after initial infection. The inflammatory response considerably thickens the stroma and neovascularization continues almost reaching the central cornea (see Fig. 35.2). Severe lesions have areas of necrosis and epithelial ulcers and uveal tissues may also be involved. The lesional T-cells, which account for only a minority of the inflammatory cells present, are mainly CD4+ T-cells. Judging from a variety of approaches, the principal cytokine necessary for the lesion expression is IFNγ (Tang and Hendricks, 1996; Deshpande et al., 2002). However, SK can still be induced in animals lacking this cytokine (Bouley et al., 1995). In cases where lesions do diminish in severity, the cytokine IL-10 is upregulated (Babu et al., 1995). Furthermore, the artificial expression of IL-10 or IL-4 early in the syndrome can markedly diminish lesions (Daheshia et al., 1998b). Such observations indicate that CD4+ Th1 are the principal aggressors but if a type 2 response can be induced, lesions will resolve. Whether such ideas can be applied usefully to the human system warrants investigation.
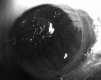
Fig. 35.2
Corneal blood vessels at the peak of HSK lesions in the mouse (day 15) with evidence of corneal opacity, necrosis and epithelial ulcers.
Very recently the severity of SK lesions was shown to be modulated by a second species of CD4+ T-cells (Suvas et al., 2004). These were CD4+CD25+ T-regulatory cells (Treg) found operative in autoimmune inflammatory lesions (Shevach, 2000). Accordingly, in animals unable to generate Treg responses, SK lesions were more severe and animals more susceptible to a low dose of infection (Suvas et al., 2004). In addition, there is evidence that the CD8 T cell response to HSV provides a protective function against SK (Mercadal et al., 1993; Gangappa et al., 1999; Banerjee et al., 2004b, 2005). The mechanisms by which Treg or CD8+ T-cells exert controlling effects on SK expression are not currently understood but are being actively explored.
A central issue in SK pathogenesis is the nature of antigens recognized by the CD4+ T-cell orchestrators and if such recognition occurs in the extra-lymphoid or lymphoid sites (or both). This issue becomes of interest since at the time when T-cells invade the cornea, replicating virus has usually disappeared (Babu et al., 1996). Moreover, certainly at the time of peak lesions (15 days), the presence of viral antigens in stromal tissues cannot be demonstrated (Babu et al., 1996). Conceivably, viral peptides expressed by DC could still be present in the cornea and draining lymph nodes, although usually T-cell target peptides turn over within 2–3 days after protein processing. Since new protein formation appears to have ceased by 6 days p.i., it is difficult to support the logical notion that peptides derived from viral proteins are the target antigens recognized by the Taggressors.
An alternative concept is that viral specific T-cells are initially responsible for the immunopathology but subsequently the chronic phase is maintained by an autoreactive response (Deshpande et al., 2002). Here the idea is that the virus infection results in unmasking of the some corneal autoantigen (see Fig. 35.3), tolerance is broken and the autoreactive T-cells induced are responsible for orchestrating lesions. A modification of this idea favored by the Cantor group is that the autoimmune process is set off by some viral peptide sharing reactivity to the unmasked corneal autoantigen (Zhao et al., 1998). Thus the initial antiviral response subsequently becomes sustained by autoreactive Taggressor cells. This concept of molecular mimicry has aroused much interest and discussion (Deshpande et al., 2002). Its best support comes from studies on closely related inbred mice. Here it would seem that the UL6 protein of HSV possesses molecular mimicry with an autopeptide that in fact represents a sequence also found on an immunoglobulin isotype (Zhao et al., 1998). The molecular mimicry idea is not accepted by other groups for a number of reasons (See Table 35.2). Most, especially the UL6 proteins of HSV appear not to induce T cell responses in animals following infection with HSV (Deshpande et al., 2001a). In humans also the UL6 protein appears not to be recognized (Verjans et al., 1998; Koelle et al., 2000; Ellison et al., 2003).
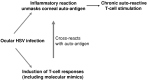
Fig. 35.3
The autoimmunity model of SK pathogenesis.
Table 35.2
Lack of evidence for molecular mimicry between HSV UL6 peptide and corneal antigen.
An alternative idea to explain how CD4+ T-cells become activated is that the inflammatory process could be initiated by viral antigen recognizing T-cells, but subsequently is maintained by cells of the effector memory phenotype that escape into the cornea because of the highly permeable neovascular bed. Such cells, in turn, become activated by inflammatory molecules initially released by viral antigen reactive cells. The responding cells, release inflammatory cytokines and so the process continues (see Fig. 35.4). This idea is supported by the observation that abundant non-antigen T-cells can be demonstrated and that it is possible to develop lesions identical to SK in animals whose T-cells are genetically incapable of recognizing viral antigens (Gangappa et al., 1998; Deshpande et al., 2001b; Banerjee et al., 2002). Such was shown in several T-cell transgenic mice on SCID or RAG–/– backgrounds whose recognition repertoire did not include HSV antigen recognition (Gangappa et al., 1998; Deshpande et al., 2001b; Banerjee et al., 2002). In these models, the chronic source of activating cytokines were cells dying of HSV infection since in this instance virus persisted and spread to the stromal site of inflammation (Gangappa et al., 1998; Deshpande et al., 2001b; Banerjee et al., 2002).

Fig. 35.4
Bystander activation model for SK pathogenesis.
Other ideas have also been advocated to explain which agonists drive SK, especially in the chronic phase, but the issue remains unresolved. The candidate agonists include PAMP expressed by virus, superantigen expression and inflammatory reactions driven by stress proteins (Deshpande et al., 2002). Currently, a favored idea is that the HSV DNA could itself be pro-inflammatory because of its high content of bioactive CpG containing deoxynucleotide motifs (Zheng et al., 2002). Such ideas await verification.
Human uveitis
Herpes simplex virus infection of the iris, ciliary body and the choroid results in an inflammatory condition known as HSV uveitis. This lesion usually accompanies SK or DK but occasionally it can stand alone. In many cases, it can occur in the absence of a previous history of HSV infection. Ophthalmoscopic examination reveals the presence of fine keratic precipitates (KP) and anterior chamber inflammation that ranges from mild to severe (Liesegang, 1999). The virus can be detected in the inflamed iris by electron microscopy. There seems to be a lack of consensus about the isolation of infectious virus from the anterior chamber of patients with HSV uveitis (Kimura, 1962; Sundmacher and Neumann-Haefelin, 1979b). This disparity may, however, result from differences among patients in the relative concentrations of anti-HSV antibodies to the virus in the anterior chamber that is needed to clear virus. Interestingly, intra-ocular antibodies recognize different viral antigens than those recognized by antibodies in the systemic immune response (Peek et al., 2002). The implications of this finding are unclear but possibly persisting intraocular antibodies could be involved in the development of secondary uveitis by the formation of toxic immune complexes. Currently, the only evidence supporting an immunopathological basis for HSV uveitis in humans is the benefit of treatment achieved with administration of topical corticosteroids along with systemic acyclovir (Liesegang, 1999). Anterior chamber uveitis can precede ARN, the latter lesion initially virologic, but later on includes immune mediated events (discussed later).
The rabbit model for HSV uveitis
The possible immunopathological nature of HSV uveitis has been revealed in studies with the rabbit model (Oh, 1976). Injection of live virus directly into the vitreous humor (intravitreal injections) of the rabbit eye results in a slowly progressing inflammation of the uveal tract. Eventually an HSV neutralizing antibody response in the infected eyes, clears the virus. Only live virus is capable of producing primary uveitis. However in eyes that have recovered, secondary uveitis can be induced by injection of inactivated virus (Oh, 1976). The disease kinetics of this secondary disease is faster than that seen with the primary infection. Such results indicate that in cases of secondary uveitis the pathogenic mechanism is likely to be immune mediated.
Acute retinal necrosis (ARN) in humans
This dramatic and devastating lesion can be caused by VZV or more commonly by HSV. Lesions can result from primary or recurrent infection and in the case of VZV can stand alone or accompany chicken pox or zoster (Ganatra et al., 2000). Without treatment the virus destroys the retina in 7–14 days. The initial stages of acute retinal necrosis (ARN) appear to be the direct result of viral damage but later stages involve immune mediated events.
Either HSV-1 or 2 can be involved. HSV-2 is usually the cause in infants, children and young adults (average age 27). In adults HSV-1 is usually the culprit (average age 50). With HSV-2 induced ARN there is often a history of encephalitis and meningitis. It is thought that virus remains in the brain after such lesions, but then passes from that site to affect the retina (Margolis and Atherton, 1996). Typically, ARN starts off as a virologic lesion, but subsequently becomes immune mediated. The vitreous fills up with inflammatory cells and occlusive vasculitis can occur which deprives the retina of its blood supply (Margolis and Atherton, 1996). In addition, both CD4+ and CD8+ T-cells, possessing cytolytic and cytokine secreting functions, have been isolated from intraocular fluids of patients. These cells have been found to be reactive with HSV UL46 and UL47 encoded tegument proteins VP11/12 and VP13/14 (Verjans et al., 2000).
In adults where HSV-1 is the usual cause, ARN is most commonly unilateral and usually associated with encephalitis. However there are two cases of retinitis that have occurred years after recovery from encephalitis (Margolis and Atherton, 1996).
The mouse model of ARN
Both the mouse and the rabbit model have been employed to study the pathogenesis of ARN and have yielded interesting clues for pathogenesis. If injected intra-cerebrally, virus spreads to the retina via the optic nerve (Atherton, 2001), providing evidence that a similar effect could occur in humans accounting for ARN long after an episode of encephalitis (Margolis and Atherton, 1996). ARN can also be induced by injecting HSV into the anterior chamber. Such infection usually fails to cause lesions in the injected eye, that is presumed to be protected by an IFNα response (Atherton, 2001). However, after a few days the contralateral eye develops a severe retinitis. This response is assumed to be immune mediated since it is of much milder extent in T-deficient mice. Moreover, reconstituting such mice with CD4+ immune T-cells, but not CD8+, restores lesion expression (Atherton, 2001).
Taken together, results from the mouse and human studies support the idea that a combination of viral infection of the retina and virus specific T-lymphocytes is likely involved in the pathogenesis of ARN.
Herpes simplex virus in the nervous system
In humans the most dramatic and devastating disease associated with HSV infection is herpes simplex encephalitis (HSE). Fortunately this is a rare syndrome for it is usually lethal or leaves patients with serious neurological damage. In adults, HSE is usually caused by HSV-1 and can occur following primary or more commonly recurrent infection (Whitley, 2001). Lesions are mainly considered to be the direct cytolytic effect of the virus. However, inflammatory reactions occur that include both CD4+ and CD8+ T-cells. In about 3% of adult cases demyelination has been noted likely a consequence of a T cell mediated immunopathological reaction (R. J. Whitley, 2003, personal communication). HSE in infants is occasionally associated with involvement of the retina (ARN).
Whereas HSE is a very rare disease in humans, heterologous hosts infected with this alphaherpesvirus are far more likely to suffer from HSE. Thus primary infection of susceptible mouse strains, especially with HSV-2, results in spread to the CNS and death from encephalitis (Hudson et al., 1991; Whitley, 2001). Whereas in humans there is no evidence of neurotropic strains of HSV, in mice some viral strains are far more neurotropic than others (LaVail et al., 1997). In some cases, the neurotropism has been associated with known amino acids in a single protein (Diefenbach et al., 2002). The rodent form of HSE largely represents a direct effect of viral destruction, but immunopathology can play a role (Kastrukoff et al., 1993; Hudson and Strelein, 1994). Multifocal brain demyelination (MBD) has been reported in susceptible mouse strains upon lip inoculation with HSV-1 and immunosuppression prevents the development of such lesions (Kastrukoff et al., 1987; Kastrukoff et al., 1993). Of the two major T-subsets, CD8+ T-cells appear to be involved in the focal lesions of the brain and depleting such cells prevents lesion development (Hudson and Streilein, 1994). Other studies indicate that CD8+ T-cells play both a protective and pathogenic role in encephalitis (Anglen et al., 2003). These studies evaluated the role of such cells in stress induced HSE. If present prior to an infection, protection ensues, possibly by limiting the HSV replication and spread within the CNS; the delayed entrance of CD8+ T-cells could result in pathology, based on limited evidence (Anglen et al., 2003).
Ganglionitis
A characteristic feature of all alphaherpesviruses is that they succeed in gaining access to sensory nerve fibers during primary infection and pass by retrograde axonal transport to the nerve cell bodies in the appropriate ganglion. At that site, whereas some neurons appear to support a productive infection, in others an alternative replication cycle is initiated that results in latency (Roizman and Knipe, 2001). With HSV, at least, latency is thought to be an immunologically cryptic situation, since the viral transcripts expressed have no protein product (Roizman and Knipe, 2001). Latency in a particular neuron can be maintained indefinitely, but some infected neurons restart the productive cycle and progeny virus spreads by anterograde transport to peripheral sites, such as the cornea. In their homologous hosts, alphaherpesviruses rarely spread to the CNS after primary infection. Such events are quite common in heterologous hosts such as HSV in the mouse. Moreover, reactivation in homologous hosts often results in recrudescent lesions, but such are rare in heterologous situations. In the mouse latently infected with HSV, occasional neurons undergo reactivation (about 1 neuron in 5 days) and this induces a notable local inflammatory reaction (Feldman et al., 2002). This likely prevents widespread dissemination in the ganglion.
Another event that characterizes HSV infection in heterologous hosts is a marked and prolonged ganglionitis that occurs after primary infection (Shimeld et al., 1995; Liu et al., 1996). This represents an immune mediated event that mainly involves CD8 + T-cells (Liu et al., 1996; Liu et al., 2000; Khanna et al., 2003). The CD8 + T-cells seemingly function to purge productively infected neurons of virus, rather than killing them by a cytotoxic mechanism (Liu et al., 2000; Khanna et al., 2003). Hence there is no tissue damage and strictly speaking no pathology. Currently, the relevance of heterologous ganglionitis is not understood nor is it known if a similar phenomenon occurs in infected human ganglia. Ganglionitis, may be another example of events that occur only in heterologous hosts infected with alphaherpesviruses.
The course of events in the mouse ganglion has been carefully studied and they tell an intriguing story (Liu et al., 1996; Liu et al., 2000; Khanna et al., 2003). The initial events involve viral replication and an inflammatory cascade that resembles that described for SK. However, after 7–10 days, CD4+ cells appear to enter and orchestrate subsequent events in SK whereas a remarkably high percentage of the inflammatory cells in the ganglia are CD8+ T-cells. Moreover, most of these are viral antigen specific and maintain this phenotype for months, which would indicate their continuous activation by antigen. However, demonstrating such antigen has proven impossible and most would agree that, after 10 days or so, virus is latent in neurons. The ganglionitis studies, however, imply that some antigen might be expressed by neurons but these are not sacrificed (Liu et al., 2000; Khanna et al., 2003); instead they are spared by the ability of the CD8+ T-cells to purge them of their offending virus (Liu et al., 2000; Khanna et al., 2003). These ideas remain to be proven and shown to be not purely a murine idiosyncrasy.
Other possible HSV induced immune mediated conditions
Herpes simplex virus affects the majority of mankind and it usually persists in some form in all it infects. Indeed, the latest sensitive molecular approaches have revealed that HSV DNA can be found in many tissues previously not recognized as an infection site. Such observations have led to the speculation that HSV could contribute to the cause of several chronic inflammatory diseases. For example, some have associated HSV with Alzheimer’s disease based on the high correlation between HSV-1 in the brain and Alzheimer’s disease (Pyles, 2001). On the same grounds, HSV-1 has been suggested to be a risk factor in other conditions like in Behcet’s disease, Bell’s palsy and Parkinson’s disease (Hegab and Al-Mutawa, 2000; Simmons, 2002; Hemling et al., 2003). Furthermore, HSV could also be a contributory cause of arteritis leading to atherosclerosis, an idea supported by some animal studies (Leinonen and Saikku, 2002).
In all cases where HSV is associated with chronic inflammatory lesions, experimental verification of a causative role is lacking.
References
- Anglen C. S., Truckenmiller M. E., Schell T. D., Bonneau R. H. 2003The dual role of CD8+ T lymphocytes in the development of stress-induced herpes simplex encephalitis J. Neuroimmunol. 140(1–2), 13–27. [PubMed: 12864968]
- Atherton S. S. Acute retinal necrosis: insights into pathogenesis from the mouse model. Herpes. 2001;8(3):69–73. [PubMed: 11867023]
- Babu J. S., Kanangat S., Rouse B. T. T cell cytokine mRNA expression during the course of the immunopathologic ocular disease herpetic stromal keratitis. J. Immunol. 1995;154(9):4822–4829. [PubMed: 7722330]
- Babu J. S., Thomas J., Kanangat S., Morrison L. A., Knipe D. M., Rouse B. T. Viral replication is required for induction of ocular immunopathology by herpes simplex virus. J. Virol. 1996;70(1):101–107. [PMC free article: PMC189793] [PubMed: 8523513]
- Banerjee K., Deshpande S., Zheng M., Kumaraguru U., Schoenberger S. P., Rouse B. T. Herpetic stromal keratitis in the absence of viral antigen recognition. Cell Immunol. 2002;219(2):108–118. [PubMed: 12576029]
- Banerjee K, Biswas P. S., Kim B, Lee S., Rouse B. T. CXCR2-/- mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J. Immunol. 2004a;172(2):1237–1245. [PubMed: 14707102]
- Banerjee K., Biswas P. S., Kumaraguru U., Schoenberger S. P., Rouse B. T. Protective and pathological roles of virus-specific and bystander CD8+ T cells in herpetic stromal keratitis. J. Immunol. 2004b;173(12):7575–7583. [PubMed: 15585885]
- Banerjee K., Biswas P. S., Rouse B. T. Elucidating the protective and pathologic T cell species in the virus-induced corneal immunoinflammatory condition herpetic stromal keratitis. J. Leukoc. Biol. 2005;77(1):24–32. [PubMed: 15496448]
- Biswas P. S., Banerjee K, Kim B., Rouse B. T. 2004Mice transgenic for IL-1 receptor antagonist protein are resistant to herpetic stromal keratitis: possible role for IL-1 in herpetic stromal keratitis pathogenesis J. Immunol. 172(6): 3736–3744. [PubMed: 15004178]
- Bouley D. M., Kanangat S., Wire W., Rouse B. T. Characterization of herpes simplex virus type-1 infection and herpetic stromal keratitis development in IFN-gamma knockout mice. J. Immunol. 1995;155(8):3964–3971. [PubMed: 7561104]
- Bouley D. M., Kanangat S., Rouse B. T. The role of the innate immune system in the reconstituted SCID mouse model of herpetic stromal keratitis. Clin. Immunol. Immunopathol. 1996;80(1):23–30. [PubMed: 8674236]
- Daheshia M., Kanangat S., Rouse B. T. Production of key molecules by ocular neutrophils early after herpetic infection of the cornea. Exp. Eye Res. 1998a;67(6):619–624. [PubMed: 9990326]
- Daheshia M., Kuklin N., Manickan E., Chun S., Rouse B. T. 1998bImmune induction and modulation by topical ocular administration of plasmid DNA encoding antigens and cytokines Vaccine 16(11–12), 1103–1110. [PubMed: 9682365]
- Deshpande S. P., Lee S., Zheng M., et al. Herpes simplex virus-induced keratitis: evaluation of the role of molecular mimicry in lesion pathogenesis. J. Virol. 2001a;75(7):3077–3088. [PMC free article: PMC114101] [PubMed: 11238834]
- Deshpande S., Zheng M., Lee S., et al. Bystander activation involving T lymphocytes in herpetic stromal keratitis. J. Immunol. 2001b;167(5):2902–2910. [PubMed: 11509638]
- Deshpande S. P., Zheng M, Lee S., Rouse B. T. 2002Mechanisms of pathogenesis in herpetic immunoinflammatory ocular lesions Vet. Microbiol. 86(1–2), 17–26. [PubMed: 11888686]
- Diefenbach R. J., Miranda-Saksena M., Diefenbach E., et al. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 2002;76(7):3282–3291. [PMC free article: PMC136023] [PubMed: 11884553]
- Dreizen N. G., Whitsett C. F., Stulting R. D. Modulation of HLA antigen expression on corneal epithelial and stromal cells. Invest. Ophthalmol. Vis. Sci. 1988;29(6):933–939. [PubMed: 3131265]
- Easty D. L., Shimeld C., Claoue C. M., Menage M. Herpes simplex virus isolation in chronic stromal keratitis: human and laboratory studies. Curr. Eye Res. 1987;6(1):69–74. [PubMed: 3030656]
- Ellison A. R., Yang L., Cevallos A. V., Margolis T. P. Analysis of the herpes simplex virus type 1 UL6 gene in patients with stromal keratitis. Virology. 2003;310(1):24–28. [PubMed: 12788627]
- Feldman L. T., Ellison A. R., Voytek C. C., Yang L., Krause P., Margolis T. P. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl Acad. Sci. USA. 2002;99(2):978–983. [PMC free article: PMC117416] [PubMed: 11773630]
- Foets B. J., van den Oord J. J., Billiau A., Damme, Missotten L. Heterogeneous induction of major histocompatibility complex class Ⅱ antigens on corneal endothelium by interferon-gamma. Invest. Ophthalmol. Vis. Sci. 1991;32(2):341–345. [PubMed: 1899654]
- Ganatra J. B., Chandler D., Santos C., Kuppermann B., Margolis T. P. Viral causes of the acute retinal necrosis syndrome. Am. J. Ophthalmol. 2000;129(2):166–172. [PubMed: 10682968]
- Gangappa S., Babu J. S., Thomas J., Daheshia M., Rouse B. T. Virus-induced immunoinflammatory lesions in the absence of viral antigen recognition. J. Immunol. 1998;161(8):4289–4300. [PubMed: 9780205]
- Gangappa S, Deshpande S. P., Rouse B. T. Bystander activation of CD4(+) T cells can represent an exclusive means of immunopathology in a virus infection. Eur. J. Immunol. 1999;29(11):3674–3682. [PubMed: 10556823]
- Hamrah P, Liu Y, Zhang Q., Dana M. R. The corneal stroma is endowed with a significant number of resident dendritic cells. Invest. Ophthalmol. Vis. Sci. 2003;44(2):581–589. [PubMed: 12556386]
- Hegab S., Al-Mutawa S. Immunopathogenesis of Behcet’s disease. Clin. Immunol. 2000;96(3):174–186. [PubMed: 10964535]
- Hemling N., Roytta M., Rinne J., et al. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann. Neurol. 2003;54(2):267–271. [PubMed: 12891684]
- Pepose, Holland, WilhelmusHolland, G. N., Tufail, A., and Jordan, C. N., (1996). Cytomegalovirus diseases. In Ocular Infection and Immunity, ed. , pp. 1088–1130. St. Louis: Mosby;
- Hudson S. J., Streilein J. W. Functional cytotoxic T cells are associated with focal lesions in the brains of SJL mice with experimental herpes simplex encephalitis. J. Immunol. 1994;152(11):5540–5547. [PubMed: 8189071]
- Hudson S. J., Dix R. D., Streilein J. W. Induction of encephalitis in SJL mice by intranasal infection with herpes simplex virus type 1: a possible model of herpes simplex encephalitis in humans. J. Infect. Dis. 1991;163(4):720–727. [PubMed: 1849158]
- Jager M. J., Atherton S., Bradley D., Streilein J. W. 1991Herpetic stromal keratitis in mice: less reversibility in the presence of Langerhans cells in the central cornea Curr. Eye Res. 10 Suppl, 69–73. [PubMed: 1650675]
- Kanangat S., Babu J. S., Knipe D. M., Rouse B. T. HSV-1-mediated modulation of cytokine gene expression in a permissive cell line: selective upregulation of IL-6 gene expression. Virology. 1996;219(1):295–300. [PubMed: 8623544]
- Kastrukoff L. F., Lau A. S., Kim S. U. Multifocal CNS demyelination following peripheral inoculation with herpes simplex virus type 1. Ann. Neurol. 1987;22(1):52–59. [PubMed: 2820296]
- Kastrukoff L. F., Lau A. S., Leung G. Y., Thomas E. E. 1993Contrasting effects of immunosuppression on herpes simplex virus type Ⅰ (HSV I) induced central nervous system (CNS) demyelination in mice J. Neurol. Sci. 117(1–2), 148–158. [PMC free article: PMC7172415] [PubMed: 8410049]
- Kaufman H. E., Kanai A., Ellison E. D. Herpetic iritis: demonstration of virus in the anterior chamber by fluorescent antibody techniques and electron microscopy. Am. J. Ophthalmol. 1971;71(2):465–469. [PubMed: 4323184]
- Khanna K. M., Bonneau R. H., Kinchington P. R., Hendricks R. L. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18(5):593–603. [PMC free article: PMC2871305] [PubMed: 12753737]
- Knipe, HowleyKieff, E. and Rickinson, A. B., (2001). Epstein–Barr virus and its replication. In Fields Virology, ed. , pp. 2511–2574. Philadelphia: Lipincott Williams and Wilkins;
- Kimura S. J. Herpes Simplex Uveitis: A clinical and experimental study. Trans. Am. Ophthalmol. Soc. 1962;60:440–470. [PMC free article: PMC1316509] [PubMed: 14032779]
- Kobayashi S., Shogi S., Ishizu M. Electron microscopic demonstration of virus particles in keratitis. Jpn J. Ophthalmol. 1972;16:247–250.
- Koelle D. M., Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clin. Microbiol. Rev. 2003;16(1):96–113. [PMC free article: PMC145296] [PubMed: 12525427]
- Koelle D. M., Reymond S. N., Chen H., et al. Tegument-specific, virus-reactive CD4 T cells localize to the cornea in herpes simplex virus interstitial keratitis in humans. J. Virol. 2000;74(23):10930–10938. [PMC free article: PMC113172] [PubMed: 11069987]
- Kumaraguru U., Rouse B. T. The IL-12 response to herpes simplex virus is mainly a paracrine response of reactive inflammatory cells. J. Leukoc. Biol. 2002;72(3):564–570. [PubMed: 12223525]
- LaVail J. H., Topp K. S., Giblin P. A., Garner J. A. Factors that contribute to the transneuronal spread of herpes simplex virus. J. Neurosci. Res. 1997;49(4):485–496. [PubMed: 9285524]
- Lee S., Zheng M., Kim B., Rouse B. T. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J. Clin. Invest. 2002a;110(8):1105–1111. [PMC free article: PMC150797] [PubMed: 12393846]
- Lee S., Zheng M., Deshpande S., Eo S. K., Hamilton T. A., Rouse B. T. IL-12 suppresses the expression of ocular immunoinflammatory lesions by effects on angiogenesis. J. Leukoc. Biol. 2002b;71(3):469–476. [PubMed: 11867684]
- Leinonen M., Saikku P. Evidence for infectious agents in cardiovascular disease and atherosclerosis. Lancet Infect. Dis. 2002;2(1):11–17. [PubMed: 11892489]
- Liesegang T. J. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea. 1999;18(2):127–143. [PubMed: 10090358]
- Liu T., Tang Q., Hendricks R. L. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol. 1996;70(1):264–271. [PMC free article: PMC189813] [PubMed: 8523535]
- Liu T., Khanna K. M., Chen X., Fink D. J., Hendricks R. L. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 2000;191(9):1459–1466. [PMC free article: PMC2213436] [PubMed: 10790421]
- Pepose J. S., Holland G. N., Wilhelmus K. R.Margolis, T. P. and Atherton, S, S., (1996). Herpes simplex virus diseases: Posterior segment of the eye. In Ocular Infection and Immunity. eds, pp. 1155–1168. St. Loius: Mosby;
- Matoba A. Y. Ocular disease associated with Epstein–Barr virus infection. Surv. Ophthalmol. 1990;35(2):145–150. [PubMed: 2173161]
- Mercadal C. M., Bouley D. M., DeStephano D., Rouse B. T. Herpetic stromal keratitis in the reconstituted scid mouse model. J. Virol. 1993;67(6):3404–3408. [PMC free article: PMC237684] [PubMed: 8098778]
- Metcalf J. F., Kaufman H. E. Herpetic stromal keratitis-evidence for cell-mediated immunopathogenesis. Am. J. Ophthalmol. 1976;82(6):827–834. [PubMed: 187060]
- Metcalf J. F., Hamilton D. S., Reichert R. W. Herpetic keratitis in athymic (nude) mice. Infect. Immun. 1979;26(3):1164–1171. [PMC free article: PMC414742] [PubMed: 160887]
- Meyers R. L., Chitjian P. A. Immunology of herpesvirus infection: immunity to herpes simplex virus in eye infections. Surv. Ophthalmol. 1976;21(2):194–204. [PubMed: 185741]
- Meyers-Elliott R. H., Pettit T. H., Maxwell W. A. Viral antigens in the immune ring of Herpes simplex stromal keratitis. Arch. Ophthalmol. 1980;98(5):897–904. [PubMed: 6246864]
- Meyers-Elliot R. H., Chitjian P. A., and Dethiefs, B. A (1983Experimental herpesvirus keratitis in the rabbit: topical versus intrastromal infection routes Ophthalmic Res. 15240–256. [PubMed: 6316227]
- Miller J. K., Laycock K. A., Nash M. M., Pepose J. S. Corneal Langerhans cell dynamics after herpes simplex virus reactivation. Invest. Ophthalmol. Vis. Sci. 1993;34(7):2282–2290. [PubMed: 8389344]
- Neta R., Sayers T. J., Oppenheim J. J. Relationship of TNF to interleukins. Immunol. Ser. 1992;56:499–566. [PubMed: 1550874]
- Niemialtowski M. G., Rouse B. T. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J. Immunol. 1992;149(9):3035–3039. [PubMed: 1357034]
- Norn M. S. Dendritic (herpetic) keratitis. I. Incidence–seasonal variations – recurrence rate–visual impairment–therapy. Acta Ophthalmol. 1970;48(1):91–107. [PubMed: 5467720]
- Oh J. O. Primary and secondary herpes simplex uveitis in rabbits. Surv. Ophthalmol. 1976;21(2):178–184. [PubMed: 185739]
- Osorio Y., Wechsler S. L., Nesburn A. B., Ghiasi H. 2002Reduced severity of HSV-1-induced corneal scarring in IL-12-deficient mice Virus Res. 90(1–2), 317–326. [PubMed: 12457985]
- Pavilack M. A., Elner V. M., Elner S. G., Todd R. F. 3rd, Huber A. R. Differential expression of human corneal and perilimbal ICAM-1 by inflammatory cytokines. Invest. Ophthalmol. Vis. Sci. 1992;33(3):564–573. [PubMed: 1347522]
- Peek R., Verjans G. M., Meek B. Herpes simplex virus infection of the human eye induces a compartmentalized virus-specific B cell response. J. Infect. Dis. 2002;186(11):1539–1546. [PubMed: 12447728]
- Pepose J. S. 1991aExternal ocular herpesvirus infections in immunodeficiency Curr. Eye Res. 10 Suppl, 87–95. [PubMed: 1864093]
- Pepose J. S. Herpes simplex keratitis: role of viral infection versus immune response. Surv. Ophthalmol. 1991b;35(5):345–352. [PubMed: 2038719]
- Pepose J. S., Nestor M. S., Gardner K. M., Foos R. Y., Pettit T. H. Composition of cellular infiltrates in rejected human corneal allografts. Graefes Arch. Clin. Exp. Ophthalmol. 1985a;222(3):128–133. [PubMed: 3884454]
- Pepose J. S., Gardner K. M., Nestor M. S., Foos R. Y., Pettit T. H. Detection of HLA class Ⅰ and Ⅱ antigens in rejected human corneal allografts. Ophthalmology. 1985b;92(11):1480–1484. [PubMed: 3909034]
- Pepose, Holland, WilhelmusPepose, J. S., Leib, D. A., Stuart, M., and Easty, D., (1996). Herpes simplex virus diseases: anterior segment of the eye. In Ocular Infection and Immunity, ed. , pp. 905–932. St. Louis: Mosby;
- Pepose J. S., Margolis T. P., LaRussa P., Pavan-Langston D. Ocular complications of smallpox vaccination. Am. J. Ophthalmol. 2003;136(2):343–352. [PubMed: 12888060]
- Pyles R. B. The association of herpes simplex virus and Alzheimer’s disease: a potential synthesis of genetic and environmental factors. Herpes. 2001;8(3):64–68. [PubMed: 11867022]
- Rager-Zisman B., Quan P. C., Rosner M., Moller J. R., Bloom B. R. Role of NK cells in protection of mice against herpes simplex virus-1 infection. J. Immunol. 1987;138(3):884–888. [PubMed: 3805719]
- Reddehase M. J. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002;2:831–844. [PubMed: 12415307]
- Remeijer L., Doornenbal P., Geerards A. J., Rijneveld W. A., Beekhuis W. H. Newly acquired herpes simplex virus keratitis after penetrating keratoplasty. Ophthalmology. 1997;104(4):648–652. [PubMed: 9111258]
- Remeijer L., Maertzdorf J., Buitenwerf J., Osterhaus A. D., Verjans G. M. Corneal herpes simplex virus type 1 superinfection in patients with recrudescent herpetic keratitis. Invest. Ophthalmol. Vis. Sci. 2002;43(2):358–363. [PubMed: 11818377]
- Knipe, HowleyRoizman, B. and Knipe, D. M., (2001). Herpes simplex viruses and their replication. In Fields Virology, ed. , pp. 2399–2460. Philadelphia: Lipincott Williams and Wilkins;
- Russell R. G., Nasisse M. P., Larsen H. S., Rouse B. T. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest. Ophthalmol. Vis. Sci. 1984;25(8):938–944. [PubMed: 6611324]
- Shevach E. M. Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 2000;18:423–449. [PubMed: 10837065]
- Shimeld C., Hill T., Blyth B., Easty D. An improved model of recurrent herpetic eye disease in mice. Curr. Eye Res. 1989;8(11):1193–1205. [PubMed: 2558849]
- Shimeld C., Whiteland J. L., Nicholls S. M. et alImmune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J. Neuroimmunol. 1995;61(1):7–16. [PubMed: 7560014]
- Shimeld C., Whiteland J. L., Nicholls S. M., Easty D. L., Hill T. J. Immune cell infiltration in corneas of mice with recurrent herpes simplex virus disease. J. Gen. Virol. 1996;77(5):977–985. [PubMed: 8609495]
- Shimeld C., Efstathiou S., and Hill T., (2001Tracking the spread of a lacZ-tagged herpes simplex virus type 1 between the eye and the nervous system of the mouse: comparison of primary and recurrent infection J. Virol. 75(11):5252–5262. [PMC free article: PMC114931] [PubMed: 11333907]
- Simmons A. 2002Clinical manifestations and treatment considerations of herpes simplex virus infection J. Infect Dis. 186Suppl 1: S71–577. [PubMed: 12353190]
- Streilein J. W., Dana M. R., Ksander B. R. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol. Today. 1997;18(9):443–449. [PubMed: 9293161]
- Su Y. H., Yan X. T., Oakes J. E., Lausch R. N. Protective antibody therapy is associated with reduced chemokine transcripts in herpes simplex virus type 1 corneal infection. J. Virol. 1996;70(2):1277–1281. [PMC free article: PMC189943] [PubMed: 8551595]
- Sundmacher R., Neumann-Haefelin D. Herpes simplex virus isolations from the aqueous humor of patients suffering from focal iritis, endotheliitis, and prolonged disciform keratitis with glaucoma. Klin. Monatsbl. Augenheilkd. 1979a;175(4):488–501. [PubMed: 548618]
- Silverstein, O’ConnorSundmacher, R. and Neumann-Haefelin, D., (1979b). Herpes simplex virus-positive and negative keratouveitis. In Immunology and Immunopathology of the Eye. ed. , pp. 225–229. New York: Masson Publishing;
- Suvas S, Azkur A. K., Kim B. S., Kumaraguru U., Rouse B. T. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J. Immunol. 2004;172(7):4123–4132. [PubMed: 15034024]
- Swanborg R. H., Whittum-Hudson J. A., Hudson A. P. 2003Infectious agents and multiple sclerosis – are Chlamydia pneumoniae and human herpes virus 6 involved? J. Neuroimmunol136(1–2), 1–8. [PubMed: 12620637]
- Tamesis R. R., Messmer E. M., Rice B. A., Dutt J. E., Foster C. S. 1994The role of natural killer cells in the development of herpes simplex virus type 1 induced stromal keratitis in mice Eye. 8(Pt 3), 298–306. [PubMed: 7958034]
- Tang Q., Hendricks R. L. Interferon gamma regulates platelet endothelial cell adhesion molecule 1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J. Exp. Med. 1996;184(4):1435–1447. [PMC free article: PMC2192815] [PubMed: 8879215]
- Thomas J., Rouse B. T. Immunopathology of herpetic stromal keratitis: discordance in CD4+ T cell function between euthymic host and reconstituted SCID recipients. J. Immunol. 1998;160:3965–3970. [PubMed: 9558104]
- Thomas J., Gangappa S., Kanangat S., Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 1997;158(3):1383–1391. [PubMed: 9013983]
- Thomas J., Kanangat S., Rouse B. T. Herpes simplex virus replication-induced expression of chemokines and proinflammatory cytokines in the eye: implications in herpetic stromal keratitis. J. Interferon Cytokine Res. 1998;18(9):681–690. [PubMed: 9781806]
- Tran M. T., Dean D. A., Lausch R. N., Oakes J. E. Membranes of herpes simplex virus type-1-infected human corneal epithelial cells are not permeabilized to macromolecules and therefore do not release IL-1alpha. Virology. 1998;244(1):74–48. [PubMed: 9581780]
- Tumpey T. M., Chen S. H., Oakes J. E., Lausch R. N. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J. Virol. 1996;70(2):898–904. [PMC free article: PMC189893] [PubMed: 8551629]
- Verjans G. M., Remeijer L., van Binnendijk, R. S. et al1998Identification and characterization of herpes simplex virus-specific CD4+ T cells in corneas of herpetic stromal keratitis patients J. Infect. Dis. 177(2):484–488. [PubMed: 9466544]
- Verjans G. M., Dings M. E., McLauchlan J., et al. Intraocular T cells of patients with herpes simplex virus (HSV)-induced acute retinal necrosis recognize HSV tegument proteins VP11/12 and VP13/14. J. Infect. Dis. 2000;182(3):923–927. [PubMed: 10950790]
- Knipe, HowleyWhitley, R. J., (2001). Herpes simplex viruses. In Fields Virology., ed. , pp. 2461–2510. Philadelphia: Lippincott Williams and Wilkins;
- Williams L. E., Nesburn A. B., Kaufman H. E. Experimental induction of disciform keratitis. Arch. Ophthalmol. 1965;73:112–118. [PubMed: 14223669]
- Yan X. T., Tumpey T. M., Kunkel S. L., Oakes J. E., Lausch R. N. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest. Ophthalmol. Vis. Sci. 1998;139(10):1854–1862. [PubMed: 9727408]
- Youinou P., Colin J., Mottier D. Immunological analysis of the cornea in herpetic stromal keratitis. J. Clin. Lab. Immunol. 1985;17(2):105–106. [PubMed: 3876445]
- Youinou P., Colin J., Ferec C. Monoclonal antibody analysis of blood and cornea T lymphocyte subpopulations in herpes simplex keratitis. Graefes Arch. Clin. Exp. Ophthalmol. 1986;224(2):131–133. [PubMed: 3485066]
- Zhao Z. S., Granucci F., Yeh L., Schaffer P. A., Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279(5355):1344–137. [PubMed: 9478893]
- Zheng M., Schwarz M. A., Lee S., Kumaraguru U., Rouse B. T. Control of stromal keratitis by inhibition of neovascularization. Am. J. Pathol. 2001a;159(3):1021–1029. [PMC free article: PMC1850467] [PubMed: 11549594]
- Zheng M., Deshpande S., Lee S., Ferrara N., Rouse B. T. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 2001b;75(20):9828–9835. [PMC free article: PMC114555] [PubMed: 11559816]
- Zheng M., Klinman D. M., Gierynska M., Rouse B. T. DNA containing CpG motifs induces angiogenesis. Proc. Natl Acad. Sci. USA. 2002;99(13):8944–8949. [PMC free article: PMC124403] [PubMed: 12060721]
- Review Evolutionary insights into the origin of innate and adaptive immune systems: different shades of grey.[Asian Pac J Allergy Immunol. 2...]Review Evolutionary insights into the origin of innate and adaptive immune systems: different shades of grey.Sirisinha S. Asian Pac J Allergy Immunol. 2014 Mar; 32(1):3-15.
- Review Protozoa: Pathogenesis and Defenses.[Medical Microbiology. 1996]Review Protozoa: Pathogenesis and Defenses.Seed JR. Medical Microbiology. 1996
- Review The evolution of immune mechanisms.[J Exp Zool B Mol Dev Evol. 2006]Review The evolution of immune mechanisms.Danilova N. J Exp Zool B Mol Dev Evol. 2006 Nov 15; 306(6):496-520.
- Review Viral Pathogenesis.[Medical Microbiology. 1996]Review Viral Pathogenesis.Baron S, Fons M, Albrecht T. Medical Microbiology. 1996
- Review [Mechanisms of innate immunity].[Postepy Hig Med Dosw (Online)....]Review [Mechanisms of innate immunity].Sochocka M, Błach-Olszewska Z. Postepy Hig Med Dosw (Online). 2005; 59:250-8.
- Immunopathological aspects of HSV infection - Human HerpesvirusesImmunopathological aspects of HSV infection - Human Herpesviruses
Your browsing activity is empty.
Activity recording is turned off.
See more...