Summary
Clinical characteristics.
Microphthalmia with linear skin defects (MLS) syndrome is characterized by unilateral or bilateral microphthalmia and/or anophthalmia and linear skin defects, usually involving the face and neck, which are present at birth and heal with age, leaving minimal residual scarring. Other findings can include a wide variety of other ocular abnormalities (e.g., corneal anomalies, orbital cysts, cataracts), central nervous system involvement (e.g., structural anomalies, developmental delay, infantile seizures), cardiac concerns (e.g., hypertrophic or oncocytic cardiomyopathy, atrial or ventricular septal defects, arrhythmias), short stature, diaphragmatic hernia, nail dystrophy, hearing impairment, and genitourinary malformations. Inter- and intrafamilial variability is described.
Diagnosis/testing.
The clinical diagnosis is established when the two major criteria (microphthalmia and/or anophthalmia and linear skin defects) are present and confirmed by identification of a pathogenic variant in COX7B, HCCS, or NDUFB11. However, persons with a molecular diagnosis of MLS syndrome in whom only one of the two major criteria was present have been reported: some show characteristic skin defects without ocular abnormalities and others show eye abnormalities without skin defects.
Management.
Treatment of manifestations: Use of a prosthesis under the guidance of an oculoplastics specialist for severe microphthalmia and anophthalmia; routine dermatologic care for significant skin lesions; treatment of seizures and/or other neurologic abnormalities by a pediatric neurologist; appropriate developmental therapies and special education as indicated for developmental delay and intellectual disability; routine care for other medical concerns when present.
Surveillance: Monitoring and follow up with ophthalmologist, dermatologist, pediatric neurologist, cardiologist, and other professionals as needed.
Genetic counseling.
MLS syndrome is inherited in an X-linked manner and is generally lethal in males. Most cases are simplex (i.e., a single occurrence in a family), but rare familial occurrences have been described. Women who are affected or have an MLS syndrome-associated pathogenic variant have a 50% chance of passing the genetic alteration to each offspring. Because male conceptuses with an MLS syndrome-associated pathogenic variant are typically nonviable, the likelihood of a live-born affected child is less than 50%. Molecular genetic testing of at-risk female relatives to determine their genetic status, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing for MLS syndrome are possible if the disease-causing genetic alteration has been identified in an affected family member.
Diagnosis
Suggestive Findings
Microphthalmia with linear skin defects (MLS) syndrome should be suspected in females with one or both major criteria especially in the presence of a family history consistent with X-linked inheritance with male lethality (see , , and ). Almost all individuals with MLS syndrome are female; however, a few affected males, typically with an XX karyotype, have been reported.
Reticulolinear scar lesions on the neck of a female age 36 years with an otherwise normal phenotype. Cytogenetic analysis revealed 46,X,del(X)(p22.3 pter) [Lindsay et al 1994].
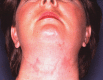
Bilateral microphthalmia and irregular linear skin areas involving the face and neck in a female infant with MLS syndrome who has a single-nucleotide variant in exon 6 of HCCS [Wimplinger et al 2006]
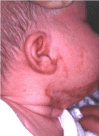
Typical linear skin lesions on the face and neck of a newborn female with MLS syndrome who has a deletion of exons 1-3 of HCCS [Morleo et al 2005, Wimplinger et al 2006]
Establishing the Diagnosis
The clinical signs observed in MLS syndrome are considered major if they are present in at least 70% of affected individuals and minor if they are less frequent (see Clinical Description, Minor Criteria).
The clinical diagnosis of MLS syndrome can be made when the two major criteria are present [al-Gazali et al 1990, Happle et al 1993]; however, persons with a molecular diagnosis of MLS syndrome in whom only one of the two major criteria was present have been reported: some show characteristic skin defects without ocular abnormalities (see ); others show eye abnormalities without skin defects [Morleo & Franco 2008].
Minor criteria in the presence of a family history consistent with X-linked inheritance with male lethality supports the clinical diagnosis of MLS syndrome.
Female proband. The diagnosis of MLS syndrome is established in a female proband by identification of a heterozygous pathogenic variant in COX7B, HCCS, or NDUFB11 on molecular genetic testing (see Table 1).
Male proband. The diagnosis of MLS syndrome is established in a male proband by identification of a hemizygous pathogenic variant in COX7B, HCCS, or NDUFB11 on molecular genetic testing (see Table 1).
Molecular testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive
genomic testing (chromosomal microarray analysis, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of MLS syndrome is broad, individuals with both major criteria are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of MLS syndrome has not been considered due to atypical findings are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of MLS syndrome, molecular genetic testing approaches can include chromosome microarray analysis or use of a multigene panel:
Chromosomal microarray analysis (CMA)
. CMA should be the first genetic test as about 90% of MLS syndrome is caused by large copy number variants (CNVs), which cannot be detected by
sequence analysis of
HCCS.
Note: (1) Deletions reported in the literature were most frequently detected by
karyotype and
FISH analysis; however, CMA is used more frequently than karyotyping in clinical practice for individuals with complex medical issues and has greater resolution and precision than a karyotype. Some complex karyotypes have been reported (e.g., 45,X[18]/46,X,der(X)(p22q21)[24]/46,X,del(X)(p22)[58] and 46,X,der(X)t(X;Y)); therefore, karyotype and/or FISH follow up may be necessary based on CMA results. (2) Apparently balanced translocations have been reported in affected individuals [
Vergult et al 2013]. In an affected person in whom other testing does not reveal a causative variant, karyotype analysis may be considered.
A multigene panel that includes
COX7B,
HCCS,
NDUFB11, and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition (if CMA is not diagnostic) while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the diagnosis of MLS syndrome is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is often performed. MLS syndrome is likely to be diagnosed by chromosome microarray (CMA), which is the best first test when multiple congenital abnormalities are present. If CMA is not diagnostic, additional genomic testing is indicated. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if CMA and exome sequencing are non-diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Note: Although not used for diagnosis, X-chromosome inactivation studies have been performed in females with MLS syndrome. Skewed X-chromosome inactivation has been detected in 21 of the 22 individuals with MLS syndrome analyzed to date [Anguiano et al 2003, Wimplinger et al 2006, Cain et al 2007, Schluth et al 2007, Wimplinger et al 2007a, Wimplinger et al 2007b, Hobson et al 2009, Steichen-Gersdorf et al 2010, Alberry et al 2011]. In all individuals the abnormal X is inactive.
Table 1.
Molecular Genetic Testing Used in MLS Syndrome
View in own window
| Gene 1, 2 | Proportion of MLS Syndrome Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 3 Detectable by Method |
|---|
| Sequence analysis 4 | Gene-targeted deletion/duplication analysis 5 | CMA 6, 7, 8 |
|---|
|
COX7B
| ~5% 9 | 3/3 10 | Unknown 11 | NA |
|
HCCS
| ~92% 9 | ~8% 12 | See footnote 13. | ~92% 14, 15 |
|
NDUFB11
| ~3% 9 | 2/2 9 | Unknown 11 | NA |
- 1.
Genes are listed alphabetically.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
- 7.
Chromosomal microarray analysis (CMA) using oligonucleotide arrays or SNP arrays. CMA designs in current clinical use target the Xp22.3 region.
- 8.
Gene-targeted deletion/duplication testing will detect deletions ranging from a single exon to the whole gene; however, breakpoints of large deletions and/or deletion of adjacent genes may not be detected by these methods.
- 9.
- 10.
- 11.
- 12.
- 13.
All large deletions reported to date are large deletions that encompass HCCS and surrounding sequence. Gene-targeted methods will detect single-exon up to whole-gene deletions; however, breakpoints of large deletions and/or deletion of adjacent genes may not be detected. Thus, the detection rate for HCCS-targeted deletion assays would be ~92%; however, the deletion may not be fully characterized.
- 14.
- 15.
Note: Deletions reported in the literature were most frequently detected by karyotype and FISH analysis; however, CMA is more commonly used than karyotyping in clinical practice for individuals with complex medical issues and has greater resolution and precision than a karyotype. Some complex karyotypes have been reported (e.g., 45,X[18]/46,X,der(X)(p22q21)[24]/46,X,del(X)(p22)[58] and 46,X,der(X)t(X;Y)), therefore, karyotype and/or FISH follow up may be necessary based on CMA results.
Clinical Characteristics
Clinical Description
Microphthalmia with linear skin defects (MLS) syndrome is characterized by unilateral or bilateral microphthalmia or anophthalmia (see ) and/or jagged skin defects on the face and neck (see ). MLS syndrome is usually lethal in males [Kono et al 1999, Kherbaoui-Redouani et al 2003, Wimplinger et al 2006, Wimplinger et al 2007a, Wimplinger et al 2007b, Kapur et al 2008, Sharma et al 2008, Hobson et al 2009, Steichen-Gersdorf et al 2010, Alberry et al 2011].
Phenotypic variability. Inter- and intrafamilial phenotypic variability has been described. The manifestations differ among affected individuals and, although most display the classic phenotype of MLS syndrome, many have only a subset of characteristic features: some show the characteristic skin defects without ocular abnormalities, whereas others have eye abnormalities without skin defects [Morleo & Franco 2008]. For example, a female with a normal phenotype except for typical MLS syndrome skin defects (see ) had an affected female fetus with anencephaly.
Major Criteria
Eye findings. Microphthalmia and/or anophthalmia, when present, are evident at birth in 81% of affected individuals (). Both microphthalmia and anophthalmia can be unilateral or bilateral.
Skin manifestations. In general, no new lesions are observed after birth and the skin defects heal variably with age, leaving minimal residual scarring. The cutaneous findings typically follow the lines of Blaschko corresponding to cell migration pathways evident during embryonic and fetal skin development, which (unlike dermatomes) do not correspond to innervation patterns. The restriction to the head and neck is thought to result from involvement of neural crest cells [al-Gazali et al 1990, Lindsay et al 1994].
Histologic skin findings.
Happle et al [1993] coined the acronym MIDAS (for microphthalmia, dermal aplasia, and sclerocornea), and argued that (in contrast to focal dermal hypoplasia) the erythematous lesions of dermal aplasia do not show herniation of fatty tissue. Subsequent histologic examination of skin biopsies of the linear, reticulated skin defects in six reported individuals yielded varied results, all confirming that dermal aplasia is not a histologic feature of MLS syndrome [Bird et al 1994, Eng et al 1994, Paulger et al 1997, Stratton et al 1998, Zvulunov et al 1998, Enright et al 2003].
Nomenclature
MLS syndrome, first described by al-Gazali et al [1990], was initially known as Gazali-Temple syndrome.
MLS syndrome appears to be the most appropriate designation for this disorder.
Happle et al [1993] coined the acronym MIDAS (for microphthalmia, dermal aplasia, and sclerocornea) for what is now known as MLS syndrome.
Prevalence
The disorder is rare; 64 individuals with a clinical diagnosis of MLS syndrome have been reported to date.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with microphthalmia with linear skin lesions (MLS) syndrome, the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Ophthalmologic examination
Dermatologic evaluation for skin lesions
Brain MRI for corpus callosum dysgenesis and other neurologic abnormalities
Developmental assessment, with further evaluation if significant delays are identified
Cardiac evaluation
Hearing evaluation, as hearing loss is observed in 8% of cases
Consideration of abdominal MRI and standard protocols for management of diaphragmatic hernia
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
The following are appropriate:
Under the guidance of an oculoplastics specialist, use of a prosthesis in severe microphthalmia and anophthalmia
Regular care by a dermatologist for individuals with significant skin lesions
Referral to a pediatric neurologist for evaluation and treatment if microcephaly, seizures, and/or other neurologic abnormalities are present
Appropriate developmental therapies and special education as indicated for developmental delay and intellectual disability
Standard care for cardiac concerns and other malformations, when present
Surveillance
Monitoring and follow up with ophthalmologist, dermatologist, pediatric neurologist, and other professionals as needed is appropriate.
Affected individuals with cardiac concerns should have regular complete evaluation at intervals determined by the cardiologist.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Microphthalmia with linear skin lesions (MLS) syndrome is inherited in an X-linked manner and is generally lethal in males.
Most affected individuals represent simplex cases (i.e., a single occurrence in a family).
Risk to Family Members
Parents of a female proband
Detailed clinical evaluation of the mother and review of the extended family history may help distinguish probands with a
de novo pathogenic variant from those with an inherited pathogenic variant.
Parents of a male proband
Live-born affected males with MLS syndrome are rare and are the result of a new chromosomal aberration (46,XX
karyotype and an X/Y
translocation).
Affected males who do not survive pregnancy may have inherited the
pathogenic variant from their mothers or may have a
de novo pathogenic variant.
Sibs of a proband. The risk to sibs depends on the genetic status of the mother.
If the mother of a
proband is affected and/or is known to have a
COX7B,
HCCS, or
NDUFB11 pathogenic variant, the chance of transmitting the pathogenic variant at conception is 50%. Because male conceptuses who inherit such a pathogenic variant are typically nonviable, the likelihood of a live-born affected child is less than 50%.
If the
proband represents a
simplex case (i.e., a single occurrence in a family) and if the
COX7B,
HCCS, or
NDUFB11 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to the sibs is slightly greater than that of the general population (though still <1%) because of the possibility of parental
germline mosaicism.
Offspring of a female proband. Women with a COX7B, HCCS, or NDUFB11 pathogenic variant have a 50% chance of transmitting the pathogenic variant to each child.
Males who inherit the
pathogenic variant will be affected, often with lethality during gestation.
Offspring of a male proband. Affected males are not known to reproduce.
Other family members. If the mother of the proband also has a pathogenic variant, her female family members may be at risk of having the pathogenic variant (and may or may not have clinical findings).
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Microphthalmia with Linear Skin Defects Syndrome: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
OMIM Entries for Microphthalmia with Linear Skin Defects Syndrome (View All in OMIM)
View in own window
|
300056 | HOLOCYTOCHROME C SYNTHASE; HCCS |
|
300403 | NADH-UBIQUINONE OXIDOREDUCTASE 1 BETA SUBCOMPLEX, 11; NDUFB11 |
|
300885 | CYTOCHROME c OXIDASE, SUBUNIT 7B; COX7B |
|
300887 | LINEAR SKIN DEFECTS WITH MULTIPLE CONGENITAL ANOMALIES 2; LSDMCA2 |
|
300952 | LINEAR SKIN DEFECTS WITH MULTIPLE CONGENITAL ANOMALIES 3; LSDMCA3 |
|
309801 | LINEAR SKIN DEFECTS WITH MULTIPLE CONGENITAL ANOMALIES 1; LSDMCA1 |
Introduction
COX7B, HCCS, and NDUFB11 encode proteins necessary for the proper functioning of the mitochondrial respiratory chain (MRC). "Canonic" mitochondrial diseases are usually characterized by postnatal organ failure rather than impaired development: in that regard microphthalmia with linear skin defects (MLS) syndrome can be defined as an unconventional mitochondrial disorder. A combined effect of mitochondrial respiration defects and enhanced cell death are hypothesized to result in the brain and eye abnormalities observed in MLS syndrome.
The three genes associated with MLS syndrome are all X-linked. Females with MLS syndrome show high inter- and intrafamilial phenotypic variability. Individuals can show the full MLS syndrome phenotype, or they can show isolated ocular manifestations, or aplastic skin areas restricted to face and neck with no additional abnormalities. Females with MLS syndrome may also show no features at all. A possible explanation for this clinical variability is the degree of skewed X chromosome inactivation observed in different tissues [Morleo & Franco 2008].
COX7B
Gene structure.
COX7B (RefSeq: NM_001866.2) has three coding exons. The gene spans 5.9 kb, and its transcribed mRNA is 456 bp. No information is available on alternative splicing.
Pathogenic variants. The clinically relevant variants reported to date include p.Gln19Ter, c.196delC, and c.41-2A>G [Indrieri et al 2012] (see Table 3).
Table 3.
COX7B Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.55C>T | p.Gln19Ter |
NM_001866.2
NP_001857.1
|
| c.196delC | p.Leu66CysfsTer48 |
| c.41-2A>G | p.Val14GlyfsTer19 |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
COX7B is ubiquitously expressed and encodes an 80-amino-acid mitochondrial protein that has been shown to be an integral component of cytochrome c oxidase (COX), the mitochondrial respiratory chain (MRC) complex IV. COX7B subunit is necessary for COX activity, COX assembly, and mitochondrial respiration [Indrieri et al 2013].
Abnormal gene product. Microphthalmia with linear skin defects (MLS) syndrome is caused by pathogenic loss-of-function variants in COX7B, which promote severe impairment of the MRC's terminal segment [Indrieri et al 2012]. Moreover, downregulation of the COX7B homolog (cox7B) in medaka fish (Oryzias latipes) results in microcephaly and microphthalmia, thus indicating an essential role for complex IV activity and MRC function in vertebrate CNS development [Indrieri et al 2013].
HCCS
Gene structure.
HCCS has seven exons, six of which are coding exons. The gene spans 11.8 kb, and its transcribed mRNA is long at 2,365 bp. No information on alternative splicing for this transcript is available (see Table 4). For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of HCCS disease-causing aberrations are large deletions involving the Xp22.2 chromosome region [Vergult et al 2013]; other reported pathogenic variants include p.Arg197Ter, p.Arg217Cys, and p.Glu159Lys, and a deletion of exons 1-3 [Wimplinger et al 2006, Wimplinger et al 2007b] (see Table 4).
Table 4.
HCCS Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.589C>T | p.Arg197Ter |
NM_005333.4
NP_005324.3
|
| c.649C>T | p.Arg217Cys |
| c.475G>A | p.Glu159Lys |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
HCCS is expressed in a wide variety of tissues and encodes a mitochondrial enzyme of 268 amino acids, the holocytochrome c-type synthase, which catalyzes the covalent attachment of heme to apocytochrome c, thereby leading to the mature form holocytochrome c [Bernard et al 2003].
The product of the HCCS-catalyzed reaction, cytochrome c, has two cellular functions: it is implicated in oxidative phosphorylation (OXPHOS), and it is released from mitochondria upon proapoptotic stimuli, thus playing an important role in caspase-dependent apoptosis [Jiang & Wang 2004].
Abnormal gene product. MLS syndrome is caused by pathogenic loss-of-function variants in HCCS. Recently it was hypothesized that deficiency of HCCS may not only cause functional deficits in OXPHOS, but may also lead to severe constraints in the process of apoptosis. Thus, loss of HCCS function may disturb the balance between necrosis and apoptosis and push cell death toward necrosis [Wimplinger et al 2006]. Necrosis bears the danger of inflammatory reactions, leading to substantial damage of neighboring cells that could be a key element in developing eye malformations as well as other MLS syndrome-specific features in affected individuals.
NDUFB11
Gene structure.
NDUFB11 spans 3 kb; its transcribed mRNA is 2,365 bp long. The gene comprises three exons; exon 2 is alternatively spliced [Petruzzella et al 2007].
Pathogenic variants. Reported pathogenic variants include p.Arg88Ter and c.402delG [van Rahden et al 2015].
Table 5.
NDUFB11 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.262C>T | p.Arg88Ter |
NM_019056.6
NP_061929.2
|
| c.402delG | p.Arg134SerfsTer3 |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The protein encoded by NDUFB11 is the 17.3-kd subunit 11 of the NADH dehydrogenase (ubiquinone) 1 beta subcomplex and is part of the MRC complex I (cI). The longer transcript encodes a 163-amino-acid (aa) protein, while the shorter one encodes a more abundant protein consisting of 153 aa. The NDUFB11 protein is indispensable for assembly of the cI membrane arm, for maturation of the cI holocomplex, and for cI-dependent mitochondrial respiration.
Abnormal gene product. MLS syndrome is caused by pathogenic loss-of-function variants in NDUFB11, resulting in defective cI assembly and activity leading to impairment of mitochondrial respiration [van Rahden et al 2015]. Moreover, reduced NDUFB11 is associated with slower cell growth and increased apoptosis [van Rahden et al 2015].
A combined effect of MRC defects and enhanced cell death has been shown to underlie the brain and eye abnormalities observed in an hccs-deficient medaka fish model [Indrieri et al 2013].