

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Frostig RD, editor. In Vivo Optical Imaging of Brain Function. 2nd edition. Boca Raton (FL): CRC Press/Taylor & Francis; 2009.

1.1. INTRODUCTION
Genetically encoded reporters of neural activity have great promise as tools for imaging brain function in vivo. Reproducible labeling of specific cell types and the continuous presence of indicators for long-term experiments are the most prominent advantages of this new methodology. It is becoming increasingly acknowledged that genetically encoded optical reporters, combined with advanced methods for recording these optical signals, will be the tool of choice for monitoring neural activity in the intact animal. Since the appearance of groundbreaking and influential descriptions of genetically encodable reporters of neuronal activity [1–4] intense efforts have been made to apply these new probes under in vivo conditions. To date, however, a relatively small number of optical reporters have proven useful for in vivo brain imaging or even in vitro neural preparations, and only a subset of these have proven useful for imaging in the intact mammalian brain. The amount of time that it has taken for these tools to mature is due to serious technical challenges in developing optical reporters that generate a sufficient signal in the intact brain, function correctly at mammalian body temperature, and throughout the life of the neuron are expressed at sufficient levels in the desired neurons without significantly altering their function. Nonetheless, many of these hurdles have been overcome in recent years, and several classes of genetically encoded activity reporters work robustly when expressed in a variety of different systems. Continued effort in developing probe and cell type–specific expression systems also promises to increase the utility and number of these reporters in the very near future.
In theory, the use of genetically encoded optical reporters of neural activity in vivo should be similar to (or less demanding than) that of classical, synthetic optical indicators. Indeed, like their synthetic counterparts, the major classes of genetically encoded reporters sense calcium or voltage, with a third class (the pHluorins) reporting synaptic vesicle cycling. Because genetically encoded reporters are typically derivatives of naturally expressed proteins such as calmodulin or ion channel subunits, the potential for interaction with endogenous proteins and enzyme substrates adds another level of complexity to experimental design and data interpretation. Thus, while many of the same technical issues seen with synthetic optical reporters apply to the use of genetically encoded probes, additional factors dependent on the specific design and mechanism of each type of indicator must also be considered when using these probes to measure and interpret neural activity in vivo.
In this chapter, we will review the design principles underlying three major classes of genetically encoded indicators—calcium sensors, reporters of transmitter release, and voltage sensors—as well as strategies for expressing these indicators in vivo. We will focus on the use of these probes in the mammalian brain, where their implementation has been the most challenging, although work in nonmammalian systems (i.e., zebrafish, Drosophila) and in in vitro preparations will be discussed in cases where this work has yielded important insights. We will then present examples using two of these sensors—synaptopHluorin (spH) and GCaMP2—to monitor sensory coding and postsynaptic processing in the mouse olfactory bulb. GCaMP2 and spH work via very different mechanisms and report distinct—though related—aspects of neural activity; consequently, each probe presents different advantages and difficulties when monitoring brain function in vivo. While the work presented here has been done primarily in the olfactory system, each of these probes has proven robust in its ability to report activity in a variety of neuronal systems, and so the principles discussed in this chapter should be generally applicable to imaging elsewhere in the brain.
1.2. DESIGN PRINCIPLES OF DIFFERENT GENETICALLY ENCODED REPORTER TYPES
Optical imaging of neuronal activity using fluorescent probes became a widely used method following the development of the calcium indicator fura-2 [5]. In brief, fura-2 combined the properties of a calcium-binding molecule (EGTA) with a bright fluorescent dye (stilbene chromophore) in a manner that caused a change in fluorescence output upon binding of calcium. Fluorescent protein (FP)-based sensors follow this inspiration. They typically involve the molecular fusion of a sensor protein that undergoes conformational transitions in response to fluctuations in a physiologic parameter (e.g., calcium concentration or membrane voltage) with a conformation-sensitive FP-based reporter protein. This approach subsequently became feasible for in vivo application with the development of bright, spectrally and optically improved variants of Aequorea victoria green FP and similar proteins from corals [6]. FP sensors have two potential advantages: they can be both genetically engineered and genetically targeted. The first feature is perhaps more practical in nature, while the ability to genetically target a fluorescent probe (that is, to stain a genetically defined cell type) is of great importance and promise for in vivo imaging.
1.2.1. Genetically Engineered FP Calcium Sensors
Förster resonance energy transfer (FRET) describes an energy transfer mechanism between two dye molecules. A donor chromophore in its excited state can transfer energy by a nonradiative, long-range dipole–dipole coupling mechanism to an acceptor chromophore in close proximity (typically <10 nm). The use of FRET to monitor distances and movements at the molecular level is now well established in biology, and the demonstration of FRET between mutational variants of GFP (Mitra et al. 1996) provided a widely used principle to transduce conformational transitions of a sensor protein in to FP optical output. The first FP calcium sensors based on FRET used the calcium-dependent interaction of the protein calmodulin with the M13 peptide as the sensor, together with pairs of blue/green or cyan/yellow FPs as reporters [4,7] (Figure 1.1A). A more recent, but similar design used the skeletal muscle calcium sensor troponin C (TnC) which, unlike calmodulin, is not expressed in neurons (Figure 1.1B). The advantage is that TnC-based sensors are proposed to avoid interactions of the sensor calmodulin with endogenous cellular mechanisms.
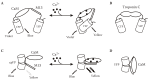
FIGURE 1.1
Genetically encoded calcium sensors: design and use for in vivo imaging. Schematics showing mechanisms of action of four genetically encoded calcium sensors: (A) yellow cameleon (from Miyawaki, A. et al., Nature, 388: 882–887, 1997); (B) TN-L15 (more...)
A second principle to “sensitize” the reporter FP to conformational alterations is circular permutation (cp). In cpFPs, the sequential order of the N- and C-terminal parts of the amino sequence are swapped, with the effect that the amino acids where the initial sequence is divided form the new N- and C-termini. Importantly, positions have been identified in the amino acid sequence of several FPs where permutation conserves the ability of the protein to form a fluorescent chromophore. The principle behind using cpFPs is that the fluorescent properties of a FP are more sensitive to the impact of a fused sensor, following structural weakening by circular permutation (Baird et al., 1999; Akemann et al., 2001) (Figures 1.1C,D). The original cpFP-based Ca2+ sensors suffered from pH sensitivity and inefficient protein folding [8–10]. However, optimizing folding efficacy of the fluorescent proteins as well as the Ca2+ affinity and dynamic range of the sensing mechanisms has subsequently yielded several generations of probes for each design principle (reviewed in Reference [11]).
Characterization of FP calcium sensors in the living mammalian (in all cases rodent) brain remains sparse, but to date CerTN-L15 (a troponin C based FRET sensor), and GCaMP2 (a green cpFP sensor) exhibit the best performance in transgenic mice [12–14]. An example of stimulus-evoked GCaMP2 signals imaged from the cerebellum of a live mouse is shown in Figure 1.1(E–J). Despite their successful use, these sensors still have weaknesses, with the TN class of sensors suffering from slow kinetics and the GCaMP2 class having a relatively low brightness [15].
1.2.2. Reporters of Transmitter Release: Phluorins
The pHluorins represent a different class of genetically encoded sensors that exploit the pH sensitivity of GFP as a signaling mechanism. “Wild-type” GFP fluorescence has a very weak pH sensitivity; in the pHluorins, this sensitivity has been enhanced through targeted mutations [2]. Several varieties of pHluorins have been generated, and their detailed functional properties are reviewed elsewhere [16–18]. “Ecliptic” pHluorins exhibit a simple reduction in fluorescence at a lower pH, while ratiometric pHluorins exhibit a shift in their excitation maximum from 395 nm at neutral pH to 475 nM at lower pH, and thus can be used—after appropriate calibration—to report absolute pH levels. The function of pHluorin as a reporter depends on the protein target to which it is linked: pHluorins can, in theory, be used to report changes in intracellular pH or movement of proteins between subcellular compartments having different pH. pHluorins are most useful for reporting neural activity when used as indicators of transmitter release. SynaptopHluorin (spH) is the most successful and widely used of the pHluorins, and consists of a “superecliptic” pHluorin linked to the luminal side of the vesicle trafficking protein VAMP-2 (Figure 1.2A) [2,16,17]. The low pH inside synaptic vesicles shifts the chromophore of the spH protein to a protonated, nonfluorescent state, causing spH fluorescence to be nearly completely eliminated (importantly, this mechanism “eclipses” photon absorption by the chromophore, and so is not a straightforward fluorescence “quenching”). Vesicle exocytosis associated with transmitter release leads to an increase in fluorescence as the pHluorin is exposed to the neutral pH of the extracellular space. As the vesicle is recycled and reacidified, fluorescence decreases to baseline levels [17,19] (Figure 1.2A).
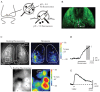
FIGURE 1.2
SynaptopHluorin as a genetically encoded reporter of transmitter release in vivo. (A) Schematic showing the mechanism of action of synaptopHluorin (spH). (B) Expression of spH in mouse olfactory receptor neurons. Image shows spH fluorescence from the (more...)
SpH has been widely used in cell culture and in in vitro preparations for studying the dynamics of vesicle recycling and, to a lesser degree, transmitter release itself [17,19–21]. It has also proven to be among the most robust of the genetically encoded indicators for in vivo or ex vivo imaging of neural activity, being effective in reporting evoked transmitter release in preparations including Drosophila olfactory sensory neurons and interneurons [22], mouse olfactory receptor neurons [23], mouse neuromuscular junction [24], and mouse hippocampal mossy fibers [25]. Examples of resting fluorescence and odorant-evoked spH signals imaged from mice expressing spH in olfactory receptor neurons is shown in Figures 1.2(B–D). The robustness of spH across systems and animal models is likely because its mechanism of action depends on a simple change in the surrounding ionic environment rather than a conformational change in its target protein. Limitations of spH include the fact that it specifically reports transmitter release, and thus is a useful indicator of “activity” only at presynaptic terminals. Its efficacy as a reporter will also depend on the reliability of transmitter release at the synapse of interest. Because of the slow time course of vesicle recycling, the temporal resolution of spH in vivo can also be limited (Figure 1.2D,E).
1.2.3. Genetically Engineered FP Voltage Sensors
FP-based probes for membrane voltage follow similar design principles as described for the FP calcium sensors. The sensor protein is derived from a voltage-sensitive protein (entire voltage-gated ion channel or a voltage sensor domain of either an ion channel or a voltage-operated, membrane-associated enzyme) and a FP reporter that is either a FP-FRET pair or a single FP. The first reported FP voltage sensor design was obtained by fusing wtGFP to the C-terminus of the Drosophila Shaker potassium channel [3]. This probe was designated FlaSh, for “fluorescent Shaker” and its design suggested that a native (not cp) FP can, in some way be “sensitized” by the surface of the lipid membrane (Figure 1.3A). The second prototypic design was based on the isolated voltage sensor domain from Kv2.1 and was termed VSFP1. Its concept was to exploit the well-established voltage-dependent conformational changes around the fourth transmembrane segment (S4) of the voltage sensor domain, in combination with a FRET FP pair [26] or a cpFP [27]. The third prototype, SPARC [28], was generated by inserting a FP between domains I and II of the rat skeletal muscle Na+ channel. Unfortunately, functional optical signals were small or undetectable in either Flare (a Kv1.4 variant of FlaSh), VSFP1, or SPARC [29]. A large, nonresponsive background fluorescence caused by poor membrane targeting of the probes was suggested as the main reason for the failure of these first-generation FP voltage sensors [29]. A second generation of FP voltage sensors overcame this limitation using the voltage sensor domain of a voltage-sensing nonion channel protein, Ci-VSP (Ciona intestinalis voltage-sensor-containing phospatase; [30] Figure 1.3B). The best variant of this second-generation FP voltage sensors is VSFP2.1. The fluorescence-voltage relationship with a V1/2 value in the physiological range of neuronal membrane fluctuations, together with relatively fast kinetics, makes VSFP2.1 a suitable candidate for optical measurements of neuronal activity, such as large synaptic potentials, action potential trains, and bistabilities in resting membrane potential (Figures 1.3D–G). In contrast to calcium imaging, VSFP2.1 can also directly monitor membrane hyperpolarization. For resolving single action potentials, a newer series of VSFPs (VSFP3s [31]) with faster kinetics may be advantageous. Generation and characterization of transgenic mice expressing VSFP2s and -3s are under way.

FIGURE 1.3
Genetically encoded optical reporters of membrane potential. (A–C) Schematics showing mechanism of action of three genetically encoded voltage sensors: (A) FLasH (from Siegel, M.S. and Isacoff, E.Y. Neuron, 19: 735–741, 1997), (B) VSFP2 (more...)
1.2.4. Monochromatic and Dual Color FP Probes
FPs come in different colors and can be utilized in either monochromatic sensors (single FP approaches) or combined to make dual-colored FRET-based sensors. Efficient use of FP probes requires an understanding of the advantages and disadvantages associated with these options. The dual-color types of the previously mentioned FP sensors respond with a fluorescence increase at one wavelength and a decrease at another wavelength when calcium concentration or membrane potential changes. These sensors can, therefore, be used for ratiometric measurements. Theoretically, ratiometric measures allow absolute calibration of the measured signals and are less affected by movements of the object. Absolute calibration in live animals is, however, usually impractical because the calibration procedures require exact subtraction of any offset fluorescence (“background”) that is usually inaccessible under conditions of in vivo imaging. Reduced sensitivity to movements is, however, a practical advantage when uncorrected background fluorescence is small. For imaging using a CCD-type camera, dual excitation or emission measurements are technically demanding, and so for this application, monochromatic approaches are preferable. Typically, monochromatic measurements (even with probes that are, in principle, suitable for dual color measurements) result in a better signal-to-noise ratio. One reason for this is that these probes are endowed with a better spectral separation between the excitation and emission wavelengths.
1.3. STRATEGIES FOR EXPRESSION OF GENETICALLY ENCODED REPORTERS IN VIVO
1.3.1. Virus Vectors
Several recombinant virus vectors have been successfully used for expression of FPs in the mature mammalian nervous system. These include adeno-associated virus (AAV), lentivirus, and herpes simplex virus (HSV) amplicon. While attempts to express FP sensors at appropriate levels and cell specificity are ongoing in several laboratories, published results are sparse. It appears that more time is required to establish the important question as to which viral vector is preferable for FP sensor expression.
1.3.2. Transgenic and Gene-Targeting Approaches
Transgenic and gene-targeted mice are time consuming and expensive to generate. However, once available, they are not only the most convenient approaches but are also more reliable reagents for study. They are convenient because such mice can be used immediately for imaging experiments with no need for preceding viral injection and incubation procedures. Specificity of labeling is also best defined in gene-targeted mice, where a specific gene product that defines the cell of interest is replaced by a FP sensor. Despite these obvious advantages, there are some limitations that must be considered: (1) there may be no gene product that is specific to the cell population of interest, (2) many gene products are driven by relative weak promoters resulting in low expression levels, and (3) gene targeting (in the form of standard “knock-in”) results in mice which have a normal gene expressed at lower levels (+/− mice) or even fully knocked out (−/− mice). Perhaps one of the most successful examples for application of knock-in techniques are mice that express spH selectively in mouse olfactory receptor neurons [23]. In these mice, the coding sequence for the receptor neuron-specific protein olfactory marker protein was replaced with that of spH via gene targeting. Imaging experiments using these mice are described below.
An excellent alternative are transgenic mice with strong cell type–specific regulatory sequences (promoters). In transgenic mice, typically several or more copies of the transgene are inserted into the genome so that screening for highly expressing lines is often successful. Good examples are mice in which the FP calcium sensor GCaMP2 is expressed under the regulatory sequences of the Kv3.1 potassium channel gene. GCaMP2 mice have proven suitable for imaging of presynaptic calcium transients in cerebellar parallel fibers (axons of granule cells) [13,14] and postsynaptic calcium signals of olfactory bulb mitral and periglomerular cells [32].
1.3.3. Electroporation In Utero
To overcome difficulties with virus vectors and the limitations of time-consuming and expensive transgenic/gene targeting approaches, electroporation in utero has proved to be a useful alternative in producing rodents expressing FPs in adult neurons [33,34].
1.3.4. Chemically Inducible Systems for Temporal Control of Reporter Expression
All of the three expression techniques described above can, in principle, be enhanced by including components for inducible expression [34]. In general, such systems consists of two components derived from the E. coli tetracycline-resistance operon. The tetracycline (TET) system uses a tetracycline-controlled transactivator that acts on a tetracycline response element (TRE) within the promoter that controls expression of the gene of interest. In principle there are two advantages in the context of FP sensor expression. Inducible systems allow the generation of mice in which FP sensor expression can be “turned on” following normal development and maturation in the absence of transgene expression. This approach has been to chosen to overcome fears that calcium buffering by FP calcium sensors may interfere with normal brain development. The TET system may also allow the use of a comparably weak but highly cell-specific promoter to drive the transactivator, while multiple copies of the TRE within a strong promoter ensure high expression levels. This idea has been pursued for FP calcium sensors, but so far the theoretical advantages have only been partially realized [35].
1.3.5. Recombinase-Based Systems for Cell Type–Specific Targeting
A second two-component system is based on recombinases such as Cre recombinase [34]. Cre recombinase catalyzes site-specific recombination of DNA between a 34–base pair recognition element (loxP sites). Cre recombinase is used to delete a segment of DNA flanked by LoxP sites (aka “floxed”). For targeted expression of FP sensors, their genes are delivered in inactive form (e.g., preceeded by a STOP codon flanked by a pair of loxP sites). When Cre recombinase is expressed in a specific cell type (using any of the above methods), the STOP codon is removed in this cell type, and the target gene (FP sensor) is expressed. Thus, this technology permits targeted expression of FP sensors in any subset of neurons for which one of the now many Cre recombinase-mice lines are available. Another promising approach involves placing the sensor gene in a lox-stop-lox cassette, which is then inserted into a virus and expressed under lox recombination as defined by a preexisting transgenic mouse with a FP sensor preceeded by a floxed STOP codon. Furthermore, Cre recombinase may be substituted, or used in combination with, other recombinases (e.g., Flp). These methods are reviewed in more detail elsewhere [34].
1.4. EXPERIMENTAL SETUP FOR IMAGING WITH GENETICALLY ENCODED REPORTERS
1.4.1. Optical Setup
Design of the experimental setup for in vivo imaging with genetically encoded fluorescent probes follows the same principles as for imaging with conventional, synthetic fluorescent indicators. Because the probes are typically expressed in only a subset of neurons in a particular brain region, resting fluorescence levels are often low relative to those seen after bulk loading of neurons with synthetic indicators such as AM-ester calcium-sensitive dyes or voltage-sensitive dyes. Resting fluorescence is also typically lower than that for the same neurons expressing GFP due to the lower resting fluorescence of the probes themselves. Thus, imaging systems that are optimized for use at low light levels are best suited for imaging with genetically encoded probes.
The authors typically use standard epifluorescence illumination to image optical signals from the surface of the brain in vivo. The system is built on the optics illumination turret of an Olympus BX51WI microscope. The illumination turret is hung from an X95 optical rail so that the entire underlying area is open. This design has proved extremely versatile, in that it allows for surgical setups, electro-physiological equipment, or even apparati for performing behavioral experiments to be positioned underneath the microscope during imaging. Images of two different configurations of this setup are shown in Figure 1.4. Custom-built “macroscopes” may also be used for epifluorescence imaging—typically, at lower cost [14,36]. However, using the standard Olympus illumination turret provides a high degree of flexibility, allowing easy interchange of filter cubes, objectives, and other components, as well as straightforward visual access to the preparation via the eyepieces. The ability to switch between filter cubes for imaging the GFP-based genetically encoded probes versus longer-wavelength synthetic indicators has been extremely useful when initially characterizing signals from novel probes, for example.

FIGURE 1.4
Examples of microscope system used for in vivo imaging in acute, chronic, and awake preparations. (left) The imaging setup configured for acute and chronic imaging in anesthetized animals. The system is built around an optics illumination turret of an (more...)
It is important that the light source used for imaging be low noise (“flutter” of less than 0.1%), especially at frequencies in the range of 1–20 Hz, as the kinetics of many of the stimulus-evoked optical signals recorded in vivo occur at this time-scale. We use a 150-W Xenon arc lamp from Opti-Quip and 150-W short-arc Xenon bulbs or a 150-W Halogen lamp (Moritex). These light sources are typically very stable, although fluctuations at 1–2 Hz and up to several percent of basal intensity can occur if power to the lamp is not properly adjusted or the bulb has aged. LED-based light sources also offer low noise and obviate the need for a shutter; these are now available from several manufacturers (e.g., Cairn Research, United Kingdom). We have found that these produce sufficient light output for use in most in vivo systems. A disadvantage with LED-based sources, however, is that switching to different excitation wavelengths for imaging different probes in the same preparation can be awkward, and only a limited range of wavelengths are available.
A low-noise CCD camera is essential for imaging signals from genetically encoded probes due to the low resting fluorescence of most probes in vivo. We use a 256 × 256 pixel, back-illuminated CCD camera from RedShirt Imaging (NeuroCCD, SM-256) or a cooled, 1600 × 1200 pixel Sensicam (PCO); both cameras have a dynamic range of at least 14 bits and a read noise of less than 30 e- at their highest frame rates (100 Hz and 30 Hz, respectively). In our experience, the pixel resolution of the CCD camera need not be high because light-scattering and biological noise in the mammalian brain limits spatial resolution with epifluorescence imaging. For most in vivo applications, frame rates of 10–100 Hz are sufficient to capture the dynamics of neuronal population activity as reflected by calcium-sensitive probes or by spH. Typical frame rates in our experiments are 25 Hz for GCaMP2 imaging and 7 Hz for spH imaging. The emergence of genetically encoded voltage sensors will hopefully allow for neuronal dynamics to be imaged at a higher temporal resolution, necessitating higher frame rates.
SpH, GCaMP2, and other genetically encoded calcium sensors are also amenable to multiphoton imaging in vivo [14,32]. Most two-photon laser scanning confocal microscopes suitable for in vivo imaging have been custom built to optimize power throughput, light collection, and scanning flexibility. Such a system is described elsewhere in this volume. In our experience it has proved extremely useful to have an epifluorescence/CCD-based imaging system in optical alignment with a two-photon scanning system, so that a relatively large region of brain tissue can be imaged with epifluorescence followed by two-photon imaging in targeted areas within this region [37]. In this way, large-scale functional maps can be acquired in concert with imaging of the underlying architecture at cellular- or subcellular-level resolution.
1.4.2. Physical Setup for In Vivo Imaging
The physical setup for imaging optical signals from genetically encoded probes in vivo is similar to that for imaging with synthetic dyes, with a key concern being minimization of artifacts related to head movement, breathing, and heartbeat. In our experiments in anesthetized animals, we fix the head by attaching a small bar to the skull with cyanoacrylate and dental cement, and fix this bar to a custom holder; the headholder is mounted to a ball joint to allow positioning of the head at oblique angles—a feature that is useful for imaging more lateral brain areas.
Several steps can be taken to reduce heartbeat and breathing artifacts. If possible, it is desirable to leave the skull intact and image through bone that has been thinned to a depth of ~100 μm using a low-speed (<1000 rpm) dental drill. Placing Ringer’s solution above the thinned bone renders it virtually transparent. If the bone and/or dura must be removed in order to perform microelectrode electrophysiology or pharmacology (for example), breathing artifacts can be reduced by elevating the head slightly above the body and by placing a coverslip atop the craniotomy window. We also commonly administer dexamethasone (2 mg/kg, SC) to reduce brain swelling in preparations requiring a craniotomy. Choice of anesthesia can affect the size of heartbeat and breathing artifacts; we have had the most success with pentobarbital, which does not cause an increase in cardiac output and generally suppresses respiratory volume. These effects, however, are accompanied by a narrow safety factor, and it is helpful to monitor EKG during the course of the experiment to detect early signs of hypoxia and to deliver oxygen if necessary.
Choice of anesthesia can also greatly affect the size of stimulus-evoked optical signals imaged with spH or calcium sensors, for several reasons. First, both evoked and spontaneous activity levels in the neurons expressing the reporter can be affected by anesthesia. Second, choice of anesthesia has a significant effect on the magnitude of intrinsic hemodynamic optical signals, the nature of which is discussed in more detail in the section below. In mice, stimulus-evoked hemodynamic signals imaged in the olfactory bulb are smallest with pentobarbital and isoflurane and largest with urethane anesthetics. Ketamine/xylazine has been undesirable as an anesthetic because of spontaneous hemodynamic signals that occur at roughly 0.5–1 Hz—a time course similar to that of many evoked signals seen with spH or GCaMP2.
1.4.3. Repeated, Chronic In Vivo Imaging
The maintained expression of genetically encoded optical probes in select neuronal populations makes these ideal tools for investigating stability and plasticity of brain activity by chronic imaging over long periods. Such imaging requires installation of an optical window over the brain region of interest that remains clear for as long as possible and does not damage the underlying tissue. It is possible to remove the overlying bone and cover the brain tissue with either agarose or silicone elastomer (e.g., Kwik-Sil, WPI) and seal this with a glass coverslip; [38,39] in our hands, however, the craniotomy remains stable and optically clear for only a fraction of windowed animals. An alternative is to thin the overlying bone and coat the thinned bone with cyanoacrylate; the cyanoacrylate renders the bone transparent and itself remains clear after hardening. With this approach, windows stay optically clear for 1–2 weeks, after which they show signs of bone regrowth and become opaque. The window can be rethinned or removed for imaging sessions at later time points. Regardless of the approach, it is important to confirm with independent methods that the windowing has not altered the underlying tissue. Figure 1.5 shows an example of odorant-evoked spH signals imaged from olfactory receptor neurons in a mouse at the time of window installation and 78 days later, after rethinning of the window. The response maps remain stable over this time period.
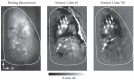
FIGURE 1.5
Long-term, chronic imaging with synaptopHluorin. (left) Resting fluorescence image of the dorsal surface of a mouse expressing spH in all olfactory receptor neurons, viewed through thinned bone. (center) Grayscale spH response map evoked by methyl valerate (more...)
1.4.4. Imaging in Awake, Head-Fixed Animals
Genetically encoded optical probes facilitate the use of imaging to monitor brain activity at high spatial and temporal resolution in awake, behaving animals. Because no labeling procedure is necessary, such imaging can be done without invasive dye-loading procedures that can disrupt behavior. Combining imaging in awake animals with repeated imaging through chronic optical windows has potential as a very powerful approach with which to investigate experience-dependent plasticity among genetically defined neuronal populations and the relationship of this activity to perception and decision making.
We have developed methods for imaging with conventional calcium-sensitive dyes in awake, head-fixed rats as they perform an operant (go/no-go) odor discrimination task; [40] the head-restraint paradigm is well established for both rats and mice [41,42] and can be easily adapted to accommodate imaging [39]. The method is essentially the same in rats or mice. Briefly, a headcap consisting of an attachment base and an inverted screw is chronically implanted onto the skull; for head-fixation, the screw is inserted through a female fitting attached to a small box or tube and the headcap tightened in place with a thumbnut. Mice or rats are mildly water deprived and given water reward during the restraint period; after 1–3 weeks of training, animals will remain head-fixed for 60–90 minutes without showing signs of stress and can be trained to perform operant behavioral tasks (typically by licking or bar press). For imaging, the fixation apparatus is positioned under a microscope objective (we use a 4×, 0.28 n.a., 30 mm working-distance objective from Olympus). The entire apparatus is mounted on XY translation plates and dual goniometers to adjust position and anterior-posterior/ medial-lateral rotation angle for optimal imaging. This setup is effective at allowing imaging during behavioral responses to sensory stimuli with minimal motion artifact. Intrinsic hemodynamic signals in the awake animal tend to be more widespread than in pentobarbital-anesthetized animals and occur both spontaneously and during stimulus-evoked activity. Methods for correcting for these artifacts are discussed below.
1.5. SIGNAL CORRECTION AND ANALYSIS METHODS
Transforming optical signals imaged with genetically encoded probes into spatial maps and temporal response patterns reflecting the underlying neural activity requires a moderate degree of signal processing. The key signal processing steps—bleaching correction, elimination of biological artifacts, and data reduction—are the same as those used for data acquired with synthetic probes, though their implementation can differ depending on the nature of the signal generated by a particular probe. At the end of this section, we also discuss methods for estimating patterns of action potential firing from the optical signals obtained in vivo.
1.5.1. Photobleaching
Imaging optical signals with epifluorescence optics typically requires some form of correction for bleaching of the probe during the course of the trial. If illumination intensities are low enough, appreciable bleaching can be avoided, obviating the need for correction. However, in our experience, the intensities needed to overcome shot-noise limitations are nearly always high enough to induce at least a moderate degree of bleaching, especially when imaging across relatively large (>1 mm2) areas using lower-magnification (and thus lower numerical aperture) objectives. The degree of photobleaching can vary with the probe itself, its expression level, its depth in the tissue, and even the anesthetic state of the animal. With spH, using a 4× (0.28 n.a.) objective covering a field of view of ~3 × 3 mm and 25% transmission of light from a 150-W Xenon arc, we typically observe bleaching of 5–10% of resting fluorescence in a single 10-s trial.
Photobleaching effects with in vivo spH imaging are somewhat unusual compared with the behavior of synthetic fluorescent reporters. First, we find that resting spH fluorescence recovers with moderate speed after bleaching, returning nearly to pretrial levels after 90 seconds [23]. Whether such recovery occurs for other genetically encoded sensors (GCaMP2, for example) is unclear, but this may be expected if the sensor protein is being continually replaced by constitutive expression or transport/diffusion from other subcellular compartments. Second, because spH fluorescence is eclipsed at the acidic pH levels within presynaptic vesicles, vesicular spH absorbs little or none of the excitation light and thus is not subject to photobleaching. Instead, bleaching of resting fluorescence arises primarily from the relatively small fraction (estimated to be ~5%) of membrane-bound spH. Thus, a reduction in resting fluorescence induced by photobleaching may not correspond to a reduction in evoked signals.
While these two features slightly complicate corrections for photobleaching—at least when imaging with spH—in practice, we have found that a simple subtraction of a “blank” (i.e., no stimulus) trial is effective at removing bleaching artifacts (Figure 1.6A–C). One concern is limiting the number of blank trials given in an imaging session, especially because the typical length of most trials for collecting in vivo data is at least several seconds. Blank trials that are distributed too sparsely lead to poor correction, since bleaching rate can change slightly during the course of an imaging session. We typically collect a blank trial approximately every 6–10 stimulus trials. The blank signal is subtracted from each stimulus trial on a pixel-by-pixel basis. To reduce the additive effects of shot noise in the blank-subtracted data, the temporally low-pass filtered prior to subtraction.

FIGURE 1.6
Bleaching and hemodynamic artifacts in fluorescence signals imaged in vivo. (A) Grayscale map of the change in fluorescence (ΔF) elicited by 2-hexanone (single trial) in a spH-expressing mouse, imaged from the dorsal olfactory bulbs through thinned (more...)
1.5.2. Hemodynamic Artifacts
Hemoglobin is a strong absorber of photons at the emission and excitation wavelengths used for imaging GFP, which is the basis for all of the current generation of genetically encoded activity probes. As a result, optical interference from blood vessels is one of the most serious limitations to imaging with these sensors in the vertebrate brain in vivo. Large surface blood vessels can obscure signals from underlying tissue. More serious, however, are artifactual signals generated by blood vessel movement. Heartbeat and respiration are one source of such artifacts, which appear most strongly at the edges of large vessels (Figure 1.6D). Another source, which can be more problematic, is activity-dependent changes in bulk blood flow to the tissue of interest; this often manifests as a widespread darkening of the tissue. This darkening is largest along major blood vessels, but also typically has a diffuse, widespread component (Figure 1.6E–G). A third source of hemodynamic artifact is spontaneous fluctuations in bulk flow that occur at frequencies of 0.2–1 Hz. All of these hemodynamic artifacts are apparent even in tissue labeled with nonactivity-dependent fluorescent markers such as Alexa Fluor 488 or GFP, and appear as fluorescence changes that can be as large as 10% of resting fluorescence—several-fold larger than evoked signals arising from the optical probe. Such artifacts are avoided in two-photon imaging, but can be a significant problem with standard epifluorescence imaging. Hemodynamic artifacts can be minimized with the correct choice of anesthesia, as described above.
Averaging signals across multiple trials can be useful in minimizing heartbeat and respiration artifact, as can triggering trial acquisition from the heartbeat and subtracting blank trials. This approach is of little help, however, in dealing with spontaneous and stimulus-evoked hemodynamic artifacts. An approach that we have found useful, however, involves applying a high-pass spatial filter to each frame of the imaged series (Figure 1.7). We use an algorithm that transforms each frame into the frequency domain and filters using a defined cutoff frequency; the cutoff frequency will vary depending on the spatial dimensions of the signal of interest. For imaging activity in glomeruli of the olfactory bulb that have an average diameter of 75–100 μm, we use a spatial cutoff frequency of 2.6 mm−1; this operation results in a ~20% reduction in glomerular response amplitudes, but a significant increase in overall signal-to-noise ratio. A particular advantage of this spatial filtering strategy is that it filters all widespread signals, regardless of time course. It can thus reduce or eliminate artifacts from photobleaching, hemodynamic signals, and even lamp noise in a single processing step. This strategy, of course, relies on the spatial dimensions of the signal of interest (i.e., that arising from the optical probe) being smaller than that of the artifactual signal—a condition that may not be satisfied in all situations. A final step that can be useful in reducing artifact from more localized hemodynamic signals (near blood vessels, for example) involves manually selecting a background region with no stimulus-evoked signal and subtracting the signal in this region from that in a nearby region of interest (Figure 1.7C).

FIGURE 1.7
Strategies for correcting for movement and intrinsic hemodynamic fluorescence signals. (A) Fluorescence signals imaged from the olfactory bulb of an awake, head-fixed rat in which olfactory receptor neurons were loaded with a synthetic calcium-sensitive (more...)
1.5.3. Green Autofluorescence Signals
Normal brain tissue exhibits “autofluorescence”; that is, fluorescence in the absence of an exogenous dye. The dominant source of green autofluorescence (in the wavelength range of GFP fluorescence excitation and emission) is the chromophore of FAD (flavin adenine dinucleotide). FAD fluorescence increases with neuronal activity and therefore green autofluorescence cannot only add background fluorescence (reducing signal-to-noise ratio) but also can contribute to stimulus-evoked optical signals recorded in the range of 500 to 600 nm. Indeed, such autofluorescence signals can be exploited as a modality of intrinsic signal imaging [43,44]. Thus, when establishing the use of FP reporter mice, it is critical to compare presumed FP signals imaged at wavelengths between 400 and 600 nm with those obtained in wild type control mice. Because evoked FAD-derived fluorescence signals typically have a slow time course, they can be easily separated from faster calcium transients monitored with probes such as GCaMP2 [14]. However, contributions from autofluorescence signals must be carefully considered for slower signals with rise times greater than approximately 200 ms (such as spH; see Figure 1.6). Contributions from autofluorescence signals become less of a concern when imaging with probes having a relatively high resting fluorescence. For example, when imaging odorant-evoked spH signals from the olfactory bulb of OMP-spH mice, resting fluorescence is 4–6 times higher than in wild-type mice; the evoked fractional fluorescence change is thus dominated by a signal from an spH signal rather than from autofluorescence. We have also found that odorant-evoked autofluorescence signals in the olfactory bulb (in wild-type mice) are much less localized than spH or GCaMP2 signals.
1.5.4. Movement Correction
With increasing interest in imaging from awake animals with conventional or genetically encoded optical probes, algorithms for offline correction of movement are becoming increasingly important. Several strategies can be implemented to correct for movement artifacts. For optical signals collected using wide-field epifluorescence, a cross-correlation-based algorithm has proved effective at correcting for relatively small movements that occur largely in the XY plane. First, a reference frame for each trial is constructed by averaging the first five imaged frames. Each subsequent frame is then high-pass filtered by subtraction with a morphologically opened frame (disk radius equals 10 pixels) and upsampled by a factor of 4 via bilinear interpolation; upsampling is necessary to correct for artifacts from movements of less than a full pixel. This step is not necessary if the original pixel resolution is high. Each upsampled frame is then cross-correlated with the reference frame; if the peak of this cross-correlation is different from the origin (indicating movement), the original frame is upsampled further (to 8× original resolution), then shifted according to the peak of its crosscorrelation with the reference. The shifted frame is then down-sampled to the original resolution.
More complex movement-correction algorithms can be implemented to correct for more serious motion artifact. For example, optical signals imaged via two-photon imaging from awake, head-fixed mice are subject to movement both within and out of the plane of focus, and the higher resolution from two-photon imaging presents a more serious challenge for movement correction. To deal with this problem, hidden Markov model-based correction algorithms have been developed that are reasonably effective at correcting for movement [39].
1.5.5. Estimating Action Potential Firing from Optical Signals
A major goal of imaging from the nervous system with optical probes is to record patterns of action potential firing. However, most genetically encoded probes used in vivo report changes in intracellular calcium concentration, which indirectly reflect action potential firing due to calcium influx through voltage-activated calcium channels. Several methods have been devised for estimating action potential firing patterns from calcium signals. The simplest method involves taking the derivative of the fluorescence signal and thresholding it to identify rapid increases in fluorescence that correspond to single spikes [45]. This approach has proved effective with high-affinity synthetic calcium indicators and in situations when individual cell bodies are visible.
A second approach for estimating action potential firing from calcium signals is not to detect single spikes, but instead to estimate changes in the relative frequency of firing of the imaged neurons by temporal deconvolution of the optical signal [46]. This approach relies on the assumption that the imaged signal is a linear convolution of the optical signal evoked by a single action potential (i.e., the impulse response) and the pattern of action potential firing. Under such conditions, the original firing pattern can be recovered from the optical signal by deconvolution with this impulse response. The impulse response can, in practice, be characterized simply as the decay rate of the calcium signal evoked by a single spike (or even burst of spikes). This decay rate can be determined by imaging from a single cell while evoking controlled spike responses via electrical stimulation or an intracellular recording electrode or while evoking responses to a white-noise-like stimulus [47]. In theory, this decay rate should be determined for each imaged neuron, which would defeat the purpose of using optical imaging as a tool for recording from many neurons. Under the appropriate conditions, however, the deconvolution method is robust enough that small differences in decay rate from neuron to neuron have little effect on estimated firing rate changes, so that the decay need only be determined for a few representative cells.
Temporal deconvolution has proved useful in estimating firing rate changes in multiple neurons imaged simultaneously using synthetic calcium indicators [46,47]. We have also used this approach to estimate temporal patterns of action potential firing at the population level, with the deconvolution applied to each group of receptor neurons converging onto a single glomerulus in the rat olfactory bulb [40]. To date, temporal deconvolution has not been applied to signals imaged with genetically encoded calcium indicators. While there are no theoretical reasons why this approach should not be applicable, limitations of the deconvolution procedure become more serious in the case of the current generation of calcium sensors. First, the response time of these sensors is somewhat slower than that of most synthetic probes due to their different mechanism of action, making it more difficult to distinguish spike-evoked calcium transients based on rise-time. Second, temporal deconvolution is valid only when the onset and decay of the optical signal varies linearly with firing rate, and there are many situations where this may not be the case—for example, if the probe nears saturation during periods of high-frequency firing, if the probe has a high threshold for detecting firing rate changes, or if intracellular calcium concentration changes independent of spiking. Most genetically encoded calcium indicators characterized to date do saturate at lower spike rates than do synthetic indicators, and are less reliable at reporting calcium influx evoked by single action potentials. Instead, a burst of several spikes occurring at relatively high-frequency is often required to evoke a detectable signal [15]. Future improvements in the design and expression of genetically encoded calcium sensors may overcome some of these limitations.
1.6. EXAMPLE EXPERIMENTS
1.6.1. SpH Imaging OF Odorant-Evoked Activity and Presynaptic Modulation of Transmitter Release
SpH has proven a sensitive and reliable reporter of stimulus-evoked activity in mouse olfactory receptor neurons [23,48,49]. Because of its mechanism of action, this probe has proved particularly useful for investigating a question of long-standing interest in olfaction: the presynaptic modulation of transmitter release from receptor neurons. Here, we present example experiments in which we use spH imaging to investigate the functional organization of this inhibition in the glomerular layer and its role in shaping odor representations in vivo. While the focus is on in vivo imaging, we have found it extremely useful to complement in vivo experiments with recordings in olfactory bulb slice preparations, which allow for more controlled stimulation paradigms and more reliable pharmacological manipulations. The slice experiments have also proved indispensable for characterizing the nature of the optical signal reported by spH; examples of these experiments are also presented here. A similar approach is used for characterizing genetically encoded optical probes that report calcium or voltage.
1.6.1.1. SpH as a Reporter of Transmitter Release
In other systems, spH has been used to track the cycling of transmitter vesicles, typically by imaging with relatively low temporal resolution and using prolonged trains of presynaptic action potentials to drive vesicle fusion and subsequent transmitter release [19, 21, 24]. However, under the right experimental conditions, spH is also an effective reporter of transmitter release (and hence synaptic transmission) when imaged at high temporal resolution and using single, brief electrical stimuli in olfactory bulb slices to elicit one or a few presynaptic action potentials. An example of one such experiment is shown in Figure 1.8. A single (0.1 ms) shock delivered to the axons of ORNs expressing spH evokes a fluorescence increase in two discrete glomeruli. The optical signal is confined to the glomerulus and is not detectable in the axons, consistent with the optical signal deriving from transmitter release at the synapse (Figure 1.8A). The shock-evoked signal has a rapid rise and a slow decay, with a time-constant of approximately 30 s (Figure 1.8B). The magnitude of the shock-evoked spH signal can be modulated by changing external calcium concentration (Figure 1.8C) or by blocking presynaptic voltage-sensitive calcium channels pharmacologically (not shown). Importantly, the dependence of the shock-evoked spH response amplitude on external [Ca2+] is nearly identical to that of EPSCs in external tufted cells, which are known to receive excitatory, monosynaptic input from ORNs (Figure 1.8D). Thus, spH is capable of reporting graded changes in the probability of transmitter release across the population of ORNs terminating with a single glomerulus.
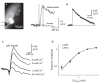
FIGURE 1.8
SynaptopHluorin as a reporter of stimulus-evoked transmitter release: in vitro validation. (A) Resting fluorescence and optical signals imaged from an olfactory bulb slice of a mouse expressing spH in olfactory receptor neurons. The position of the stimulating (more...)
Nonetheless, high temporal resolution imaging of shock-evoked spH signals reveals that the rise-time of the response (approximately 30 ms) is considerably slower than the influx of calcium into the presynaptic terminal, which can be imaged in the same experiment using synthetic, long-wavelength calcium-sensitive dyes loaded into ORNs (Figure 1.9A). In addition, close inspection of the evoked spH signal reveals a small and brief decrease in fluorescence whose origin is unclear (Figure 1.9B). The explanation for both the prolonged rise-time and the initial fluorescence lies in the mechanism of action of spH, which is to report changes in pH surrounding the fluorophore, regardless of whether or not these occur as a result of vesicle cycling. Because the majority of the spH resting fluorescence arises from the small fraction of protein present in the plasma membrane and not sequestered inside vesicles, even small changes in extracellular pH can result in significant fluorescence changes that do not directly reflect transmitter release. In the case of olfactory nerve stimulation, nerve shock elicits synchronous release from many (up to several thousand) axons in the glomerulus, which can transiently acidify the extracellular space. Indeed, increasing the buffering capacity of the extracellular solution by adding 20 mM HEPES eliminates the transient fluorescence decrease (Figure 1.9C). Likewise, activation of glutamate transporters after the response can transiently alkalinize the extracellular space because protons are cotransported during glutamate reuptake [50], causing a slower transient fluorescence increase. Blocking glutamate reuptake with TBOA eliminates this increase (Figure 1.9D). Finally, shock-evoked spH signals measured in the presence of both 20 mM HEPES and TBOA reveal fluorescence increases that are monophasic and have a rise time of less than 10 ms, as expected for transmitter release (Figure 1.9E). These experiments illustrate the importance of thoroughly characterizing the nature of the optical signal being generated by genetically encoded optical probes, especially those that are not direct reporters of membrane potential. Nonetheless, in the case of spH, despite the contribution of “artifactual” sources to the evoked optical signal, the fact that evoked response amplitudes scale linearly with EPSC amplitudes and that spH rise-time is independent of amplitude suggest that changes in evoked spH signal amplitude will accurately reflect modulation in transmitter release from the presynaptic terminal. This relationship appears to approximately hold using odorant stimulation in vivo, as evidenced by a rough correspondence between maps of odorant-evoked spH signals and those imaged with calcium-sensitive dye in the same experiment (Figure 1.10). However, we have not tested the degree to which pH-dependence or other effects may distort the dynamics of responses evoked during odorant stimulation. Thus, in our in vivo experiments, we have limited our interpretation of spH signals to focus on modulation of the response by drug application or odorant-evoked inputs to neighboring glomeruli.
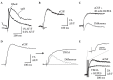
FIGURE 1.9
pH-dependent artifacts affecting stimulus-evoked spH fluorescence signals. (A) Traces showing “high-affinity” rhod dextran signals reflecting shock-evoked presynaptic calcium influx (rhod) and spH signals reflecting transmitter release (more...)
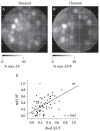
FIGURE 1.10
Correspondence between spH and presynaptic calcium signals imaged in vivo. (A) Grayscale map of the spH signal evoked by the odorant hexanal in a mouse in which olfactory receptor neurons were also loaded with the long-wavelength synthetic calcium indicator (more...)
1.6.1.2. Using SpH to Probe the Organization of Presynaptic Inhibition In Vivo
We have used spH imaging to probe the organization of presynaptic inhibition in the glomerular layer of the olfactory bulb (OB) and its role in shaping odor representations in vivo. Experiments performed in OB slice preparations using paired electrical stimulation of ORN axons reveal a strong paired-pulse suppression of transmitter release that is partially relieved by blocking ionotropic glutamatergic receptors [49]. These experiments demonstrate that ORNs—which are glutamatergic—activate post-synaptic olfactory bulb neurons that then presynaptically inhibit release in response to subsequent inputs. GABA-ergic interneurons in the glomerular layer play a major role in this inhibition: GABAB receptors are expressed on the presynaptic terminals of ORNs, and GABAB antagonists are as effective as glutamatergic antagonists at relieving paired-pulse suppression [48,49,51]. To investigate the role of this GABAB-mediated presynaptic inhibition in odor coding, we have performed several types of experiments using spH signals as measures of odorant-evoked transmitter release from ORNs in vivo [48]. In the first, we applied a GABAB antagonist (CGP35348) directly to the dorsal surface of the mouse OB while imaging odorant-evoked spH signals. In these experiments, mice were anesthetized with pentobarbital and the dura removed from the dorsal OB prior to data collection. Dura removal (when done carefully) has no negative impact on imaged responses, and in fact slightly improves the quality of imaged response maps. SpH signals remain stable or decrease slightly when control-mouse Ringer’s is applied to the OB. In contrast, when GABAB agonist baclofen is applied, odorant-evoked spH signals are greatly decreased (Figure 1.11A). The effect of baclofen can be reversed by applying CGP35348, a GABAB receptor antagonist. Applying CGP35348 alone causes a strong increase in odorant-evoked spH signals, with an average increase of approximately 80% in ΔF after 4 s of odorant presentation (Figure 1.11B). This indicates that presynaptic inhibition mediated by GABAB receptors regulates the magnitude of sensory input to the olfactory bulb.
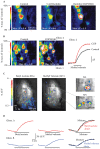
FIGURE 1.11
Imaging the in vivo regulation of receptor input to the olfactory bulb by GABAB-mediated presynaptic inhibition. (A) Pseudocolor maps of the spH signal evoked by the odorant butyl acetate. The GABAB receptor agonist baclofen reduces the amplitude of the (more...)
The localization of the spH signal to the sites of transmitter release (i.e., within glomeruli) allowed us to compare overall spatial patterns of ORN input to glomeruli before and after GABAB receptor blockade. Normalizing evoked response maps to their peak response magnitude indicates that relative patterns are unchanged by CGP35348—an observation supported by correlation analyses of response maps [48]. In other words, blocking presynaptic inhibition does not recruit input to glomeruli not initially activated by the odorant. This result also provides evidence that the odorant-evoked spH signal responds approximately linearly across much of the dynamic range of receptor inputs to glomeruli; if the spH signal had a significant threshold for reporting presynaptic activity or if it saturated at relatively low levels of activation, increasing transmitter release with CGP35348 would be expected to significantly alter relative response maps imaged with the probe. To more directly test whether presynaptic inhibition alters relative patterns of input to glomeruli—for example, via lateral inhibition between glomeruli—we used odorants chosen to selectively activate adjacent glomeruli. The rationale for this experiment is that if lateral inter-glomerular inhibition modulates ORN input in vivo, then the odorant-evoked spH signal in a glomerulus should be suppressed when its neighbor is coactivated by a different odorant. An example experiment is shown in Figure 1.11C. In this example, butyl acetate evokes strong input to a single glomerulus (the “test glomerulus”) in the central dorsal OB and weaker input to additional glomeruli. A different odorant, methyl valerate, evokes strong input to three glomeruli surrounding the test glomerulus, but evokes little or no input to the test glomerulus itself. We then asked whether coactivation of the surrounding glomeruli by methyl valerate suppressed the response of the test glomerulus to butyl acetate by presenting the two odorants as a binary mixture. As shown in Figure 1.11D, the test glomerulus’s response to the mixture was identical in both amplitude and time course to the response to butyl acetate alone. These experiments suggest that a major role of presynaptic inhibition in vivo is to regulate the strength of sensory input to the brain while maintaining the spatial maps of glomerular activation that are thought to encode odorant identity. However, several questions about the role of presynaptic inhibition in vivo are not easily addressed using spH—chiefly because of its limited temporal resolution. For example, it is unclear from these experiments whether inputs are modulated by feedback inhibition—which is apparent in slices—or via tonic inhibition under the control of (for example) centrifugal inputs. Presynaptic indicators with higher temporal resolution—such as genetically encoded or synthetic calcium indicators—would be useful in addressing this question.
1.6.2. Imaging Postsynaptic Odor Representations with GCaMP2
To date there have been few examples of genetically encoded calcium sensors used to report neural activity in the in vivo mammalian brain. One successful example is a line of transgenic mice expressing GCaMP2 under the control of the Kv 3.1 potassium channel promoter [13,14]. Certain lines of these mice show stable expression of GCaMP2 in cerebellum, pyramidal neurons throughout the neocortex, and in mitral cells and some interneurons of the olfactory bulb. The mouse lines with strong expression in the olfactory bulb can thus be used to study postsynaptic odor representations in vivo [32]. Here, we show an example of odorant-evoked GCaMP2 signals imaged from the dorsal OB in a pentobarbital-anesthetized mouse (Figure 1.12).
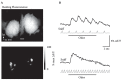
FIGURE 1.12
Postsynaptic odorant-evoked GCaMP2 signals imaged from the olfactory bulb in vivo. (A) Top: Resting fluorescence from the dorsal olfactory bulbs of a mouse expressing GCaMP2 under the control of the Kv 3.1 promoter. Bottom: Grayscale response map evoked (more...)
In this example, odorants were applied while artificially “sniffing” air through the nasal cavity via a tube inserted into the nasopharynx (a second tube directed toward the lungs permitted free respiration). Note that the resting fluorescence does not reveal clear glomeruli (Figure 1.12A), as is evident in the OMP-spH mice. This is partially due to the low fluorescence of GCaMP2 at resting calcium concentration and also to the expression in some interneurons whose processes cross glomerular boundaries. Odorant presentation evokes optical signals that have a relatively rapid rise-time as well as a relatively rapid decay; these temporal dynamics are fast enough to allow responses to each individual inhalation to be tracked—at least when presented at 1 Hz (Figure 1.12B). Thus, the temporal resolution of the GCaMP2 signal is higher than that of the spH signal and appears similar to that of synthetic calcium-sensitive dyes. Spatial maps of the evoked signal are spatially organized and appear to consist of discrete glomeruli in locations expected from separate imaging experiments using spH or synthetic dye imaging from ORNs. Finally, responses evoked during high-frequency sniffing of odorant show a lack of modulation by each sniff and instead show clear attenuation of the response during prolonged stimulation (Figure 1.12B). This frequency-dependent attenuation is also consistent with our earlier observations imaging presynaptic calcium signals [40,52]. Thus, GCaMP2 imaging allows the testing hypotheses about how sensory inputs are transformed by synaptic processing. It will, of course, be extremely interesting to combine the postsynaptic GCaMP2 imaging with imaging of presynaptic activity in the same preparation—either using spH or longer-wavelength, synthetic calcium-sensitive dye loaded into ORNs.
1.7. DISCUSSION AND FUTURE DIRECTIONS
The properties of the current generation of the major types of genetically encoded FP-based probes that are most advanced for in vivo applications are summarized in Table 1.1. The spH probes represent the only class that is currently more suitable for in vivo imaging of activity than its conventional (“synthetic,” not genetically encoded) analog (FM dyes—the synthetic analog of spH—are not suitable for in vivo experimentation). Synthetic versions of the other two classes—calcium and voltage sensors—have undergone many more person-years of development, and currently surpass genetically encoded sensors in signal-to-noise ratio, response times, linear dynamic range, range of affinities, and range of available imaging wavelengths. In particular, small molecule synthetic calcium indicators are readily applicable in vivo and continue to facilitate many invaluable insights into nervous system function. Nonetheless, genetically encoded activity calcium-sensitive FPs uniquely enable experiments in which cell type–specific and long-lasting imaging approaches are essential. Thus, continued effort in improving probe performance, as well as in developing expression strategies, should remain a high priority.
TABLE 1.1
Functional Characteristics of Genetically Encoded FP Sensors
VSFPs have not been sufficiently explored in vivo to directly compare them with available conventional voltage imaging methods. Because unselective labeling of plasma membranes by synthetic voltage probes has prohibitive effects on both the signal to noise ratio and interpretation of signals, VSFPs have the potential to overcome many of the current limitations of voltage imaging based on organic voltage sensitive dyes. Because current VSFPs have relatively modest AF/F values, however, and because S/NR is directly proportional to ΔF/F, improving this parameter will greatly enhance the prospects of these probes.
One area of improvement that has received relatively little effort to date is the development of longer-wavelength FP probes. The main advantages of excitation with and emission of longer wavelength light are (1) reduced interference with blue and green fluorescence signals from endogenous chromophores (NAD, FAD), (2) reduced interference from activity dependent changes in hemoglobin and cytochrome oxidase absorption, and (3) improved tissue penetration due to an increased “mean free path” (the average distance between scattering events) [53]. The increase in signal-to-noise ratio when imaging with longer-wavelength probes in vivo in the mammalian brain—particularly when imaging with epifluorescence—can be an order of magnitude; thus, developing longer-wavelength versions of the current generation of FP sensors could be the most efficient way to increase probe performance. Red-shifted FP calcium and voltage sensors are currently being developed in several labs, but are not yet widely distributed. Additional effort on improving probe performance should focus on increasing the kinetics as well as brightness of genetically encoded probes—in particular, calcium sensors. Fortunately, the increased interest in making genetically encoded probes useful for monitoring activity in the intact brain should help to fulfill the promise of these tools for studying nervous system function in the near future.
Finally, in this review we have focused on genetically encoded probes to optically image brain activity in vivo. During recent years a variety of techniques that allow optical and genetically targeted control of neuronal activity have emerged [54–56]. We envisage that in the near future, genetically encoded tools will allow for optical probing combined with and complemented by optical control of assemblies of specific individual nerve cells in vivo.
ACKNOWLEDGMENTS
The authors would like to thank present and past collaborators for their valuable contributions to the work described here, including J. McGann, N. Pírez, J. Verhagen, R. Carey, M.C. Cheung, D. Wesson, H. Mutoh, D. Dimitrov, W. Akemann, A. Perron, and Y. Iwamoto. We thank L. Cohen for essential guidance in methodological development. Our respective laboratories have been supported by grants from the National Institutes of Health (MW and TK), Boston University (MW), and RIKEN BSI (TK).
REFERENCES
- 1.
- Miesenbock G, Rothman JE. Patterns of synaptic activity in neural networks recorded by light emission from synaptolucins. Proc Natl Acad Sci USA. 1997;94:3402–3407. [PMC free article: PMC20382] [PubMed: 9096406]
- 2.
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. [PubMed: 9671304]
- 3.
- Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19:735–741. [PubMed: 9354320]
- 4.
- Miyawaki A, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. [PubMed: 9278050]
- 5.
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed: 3838314]
- 6.
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. [PubMed: 16614209]
- 7.
- Romoser VA, Hinkle PM, Persechini A. Detection in living cells of Ca2+−dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J Biol Chem. 1997;272:13270–13274. [PubMed: 9148946]
- 8.
- Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. [PMC free article: PMC18018] [PubMed: 10500161]
- 9.
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. [PubMed: 11175727]
- 10.
- Akemann W, Raj CD, Knöpfel T. Functional characterization of permuted enhanced green fluorescent proteins comprising varying linker peptides. Photochem Photobiol. 2001;74:356–363. [PubMed: 11547577]
- 11.
- Knöpfel T, Diez-Garcia J, Akemann W. Optical probing of neuronal circuit dynamics: Genetically encoded versus classical fluorescent sensors. Trends Neurosci. 2006;29:160–166. [PubMed: 16443289]
- 12.
- Heim N, et al. Improved calcium imaging in transgenic mice expressing a troponin C-based biosensor. Nat Methods. 2007;4:127–129. [PubMed: 17259991]
- 13.
- Diez-Garcia J, et al. Activation of cerebellar parallel fibers monitored in transgenic mice expressing a fluorescent Ca2+ indicator protein. Eur J Neurosci. 2005;22:627–635. [PubMed: 16101744]
- 14.
- Diez-Garcia J, Akemann W, Knöpfel T. In vivo calcium imaging from genetically specified target cells in mouse cerebellum. Neuroimage. 2007;34:859–869. [PubMed: 17161628]
- 15.
- Mao T, O’Connor DH, Scheuss V, Nakai J, Svoboda K. Characterization and subcellular targeting of GCaMP-type genetically encoded calcium indicators. PLoS ONE. 2008;3:e1796. [PMC free article: PMC2262942] [PubMed: 18350138]
- 16.
- Yuste R, Miller RB, Holthoff K, Zhang S, Miesenbock G. Synapto-pHluorins: Chimeras between pH-sensitive mutants of green fluorescent protein and synaptic vesicle membrane proteins as reporters of neurotransmitter release. Methods Enzymol. 2000;327:522–546. [PubMed: 11045007]
- 17.
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. [PMC free article: PMC1301110] [PubMed: 11023924]
- 18.
- Miesenbock G. A Practical Guide: Synapto-pHluorins—Genetically encoded reporters of synaptic transmission. In: Yuste R, Konnerth A, editors. Imaging in Neuroscience and Development: A Laboratory Manual. Cold Spring Harbor Press; Cold Spring Harbor, New York: 2005. pp. 599–604. [PubMed: 22301651]
- 19.
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. [PubMed: 10783237]
- 20.
- Gandhi SP, Stevens CF. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. [PubMed: 12789331]
- 21.
- Li Z, et al. Synaptic vesicle recycling studied in transgenic mice expressing synap-topHluorin. Proc Natl Acad Sci USA. 2005;102:6131–6136. [PMC free article: PMC1087931] [PubMed: 15837917]
- 22.
- Ng M, et al. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron. 2002;36:463–474. [PubMed: 12408848]
- 23.
- Bozza T, McGann JP, Mombaerts P, Wachowiak M. In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron. 2004;42:9–21. [PubMed: 15066261]
- 24.
- Wyatt RM, Balice-Gordon RJ. Heterogeneity in synaptic vesicle release at neuro-muscular synapses of mice expressing synaptopHluorin. J Neurosci. 2008;28:325–335. [PMC free article: PMC6671144] [PubMed: 18171949]
- 25.
- Suyama S, Hikima T, Sakagami H, Ishizuka T, Yawo H. Synaptic vesicle dynamics in the mossy fiber-CA3 presynaptic terminals of mouse hippocampus. Neurosci Res. 2007;59:481–490. [PubMed: 17933408]
- 26.
- Sakai R, Repunte-Canonigo V, Raj CD, Knöpfel T. Design and characterization of a DNA-encoded, voltage-sensitive fluorescent protein. Eur J Neurosci. 2001;13:2314–2318. [PubMed: 11454036]
- 27.
- Knöpfel T, Tomita K, Shimazaki R, Sakai R. Optical recordings of membrane potential using genetically targeted voltage-sensitive fluorescent proteins. Methods. 2003;30:42–48. [PubMed: 12695102]
- 28.
- Ataka K, Pieribone VA. A genetically targetable fluorescent probe of channel gating with rapid kinetics. Biophys J. 2002;82:509–516. [PMC free article: PMC1302490] [PubMed: 11751337]
- 29.
- Baker BJ, et al. Three fluorescent protein voltage sensors exhibit low plasma membrane expression in mammalian cells. J Neurosci Methods. 2007;161:32–38. [PubMed: 17126911]
- 30.
- Dimitrov D, et al. Engineering and characterization of an enhanced fluorescent protein voltage sensor. PLoS ONE. 2007;2:e440. [PMC free article: PMC1857823] [PubMed: 17487283]
- 31.
- Lundby A, Mutoh H, Dimitrov D, Akemann W, Knöpfel T. Engineering of a genetically encodable fluorescent voltage sensor exploiting fast Ci-VSP voltage-sensing movements. PLoS ONE. 2008;3:e2514. [PMC free article: PMC2429971] [PubMed: 18575613]
- 32.
- Chaigneau E, et al. The relationship between blood flow and neuronal activity in the rodent olfactory bulb. J Neurosci. 2007;27:6452–6460. [PMC free article: PMC6672435] [PubMed: 17567806]
- 33.
- Navarro-Quiroga I, Chittajallu R, Gallo V, Haydar TF. Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J Neurosci. 2007;27:5007–5011. [PMC free article: PMC2710030] [PubMed: 17494686]
- 34.
- Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. [PMC free article: PMC2628815] [PubMed: 18341986]
- 35.
- Hasan MT, et al. Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol. 2004;2:e163. [PMC free article: PMC423138] [PubMed: 15208716]
- 36.
- Ratzlaff EH, Grinvald A. A tandem-lens epifluorescence macroscope: Hundred-fold brightness advantage for wide-field imaging. J Neurosci Methods. 1991;36:127–137. [PubMed: 1905769]
- 37.
- Wachowiak M, Denk W, Friedrich RW. Functional organization of sensory input to the olfactory bulb glomerulus analyzed by two-photon calcium imaging. PNAS. 2004;101:9097–9102. [PMC free article: PMC428479] [PubMed: 15184670]
- 38.
- Trachtenberg JT, et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. [PubMed: 12490942]
- 39.
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. [PMC free article: PMC2268027] [PubMed: 17920014]
- 40.
- Verhagen JV, Wesson DW, Netoff TI, White JA, Wachowiak M. Sniffing controls an adaptive filter of sensory input to the olfactory bulb. Nat Neurosci. 2007;10:631–639. [PubMed: 17450136]
- 41.
- Boyden ES, Raymond JL. Active reversal of motor memories reveals rules governing memory encoding. Neuron. 2003;39:1031–1042. [PubMed: 12971901]
- 42.
- Katz DB, Simon SA, Nicolelis MA. Dynamic and multimodal responses of gustatory cortical neurons in awake rats. J Neurosci. 2001;21:4478–4489. [PMC free article: PMC6762775] [PubMed: 11404435]
- 43.
- Shibuki K, et al. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. [PMC free article: PMC2342977] [PubMed: 12730344]
- 44.
- Coutinho V, Mutoh H, Knöpfel T. Functional topology of the mossy fibre-granule cell—Purkinje cell system revealed by imaging of intrinsic fluorescence in mouse cerebellum. Eur J Neurosci. 2004;20:740–748. [PubMed: 15255984]
- 45.
- Smetters D, Majewska A, Yuste R. Detecting Action potentials in neuronal populations with calcium imaging. Methods. 1999;18:215–221. [PubMed: 10356353]
- 46.
- Yaksi E, Friedrich RW. Reconstruction of firing rate changes across neuronal populations by temporally deconvolved Ca(2+) imaging. Nat Methods. 2006;3:377–383. [PubMed: 16628208]
- 47.
- Ramdya P, Reiter B, Engert F. Reverse correlation of rapid calcium signals in the zebrafish optic tectum in vivo. J Neurosci Methods. 2006;157:230–237. [PubMed: 16765450]
- 48.
- McGann JP, et al. Odorant representations are modulated by intra- but not interglomerular presynaptic inhibition of olfactory sensory neurons. Neuron. 2005;48:1039–1053. [PubMed: 16364906]
- 49.
- Wachowiak M, et al. Inhibition of olfactory receptor neuron input to olfactory bulb glomeruli mediated by suppression of presynaptic calcium influx. J Neurophysiol. 2005;94:2700–2712. [PMC free article: PMC1282456] [PubMed: 15917320]
- 50.
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. [PubMed: 8857541]
- 51.
- Aroniadou-Anderjaska V, Zhou F.-M, Priest CA, Ennis M, Shipley MT. Tonic and synaptically evoked presynaptic inhibition of sensory input to rat olfactory bulb via GABAB heteroreceptors. J Neurophysiol. 2000;84:1194–1203. [PubMed: 10979995]
- 52.
- Spors H, Wachowiak M, Cohen LB, Friedrich RW. Temporal dynamics and latency patterns of receptor neuron input to the olfactory bulb. J Neurosci. 2006;26:1247–1259. [PMC free article: PMC6674558] [PubMed: 16436612]
- 53.
- Oheim M, Beaurepaire E, Chaigneau E, Mertz J, Charpak S. Two-photon microscopy in brain tissue: Parameters influencing the imaging depth. J Neurosci Methods. 2001;111:29–37. [PubMed: 11574117]
- 54.
- Miesenbock G, Kevrekidis IG. Optical imaging and control of genetically designated neurons in functioning circuits. Annu Rev Neurosci. 2005;28:533–563. [PubMed: 16022604]
- 55.
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. [PubMed: 16116447]
- 56.
- Knöpfel T. Expanding the toolbox for remote control of neuronal circuits. Nat Methods. 2008;5:293–295. [PubMed: 18376391]
- 57.
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci USA. 2001;98:3197–3202. [PMC free article: PMC30630] [PubMed: 11248055]
- INTRODUCTION
- DESIGN PRINCIPLES OF DIFFERENT GENETICALLY ENCODED REPORTER TYPES
- STRATEGIES FOR EXPRESSION OF GENETICALLY ENCODED REPORTERS IN VIVO
- EXPERIMENTAL SETUP FOR IMAGING WITH GENETICALLY ENCODED REPORTERS
- SIGNAL CORRECTION AND ANALYSIS METHODS
- EXAMPLE EXPERIMENTS
- DISCUSSION AND FUTURE DIRECTIONS
- ACKNOWLEDGMENTS
- REFERENCES
- Review Engineering Aspects of Olfaction.[Neuromorphic Olfaction. 2013]Review Engineering Aspects of Olfaction.Persaud KC. Neuromorphic Olfaction. 2013
- Review In Vivo Observations of Rapid Scattered Light Changes Associated with Neurophysiological Activity.[In Vivo Optical Imaging of Bra...]Review In Vivo Observations of Rapid Scattered Light Changes Associated with Neurophysiological Activity.Rector DM, Yao X, Harper RM, George JS. In Vivo Optical Imaging of Brain Function. 2009
- Imaging voltage in zebrafish as a route to characterizing a vertebrate functional connectome: promises and pitfalls of genetically encoded indicators.[J Neurogenet. 2016]Imaging voltage in zebrafish as a route to characterizing a vertebrate functional connectome: promises and pitfalls of genetically encoded indicators.Kibat C, Krishnan S, Ramaswamy M, Baker BJ, Jesuthasan S. J Neurogenet. 2016 Jun; 30(2):80-8.
- Using Genetically Encoded Voltage Indicators (GEVIs) to Study the Input-Output Transformation of the Mammalian Olfactory Bulb.[Front Cell Neurosci. 2019]Using Genetically Encoded Voltage Indicators (GEVIs) to Study the Input-Output Transformation of the Mammalian Olfactory Bulb.Storace DA, Cohen LB, Choi Y. Front Cell Neurosci. 2019; 13:342. Epub 2019 Jul 31.
- Review Imaging different cell populations in the mouse olfactory bulb using the genetically encoded voltage indicator ArcLight.[Neurophotonics. 2024]Review Imaging different cell populations in the mouse olfactory bulb using the genetically encoded voltage indicator ArcLight.Leong LM, Storace DA. Neurophotonics. 2024 Jul; 11(3):033402. Epub 2024 Jan 17.
- Optical Imaging of Brain Activity In Vivo Using Genetically Encoded Probes - In ...Optical Imaging of Brain Activity In Vivo Using Genetically Encoded Probes - In Vivo Optical Imaging of Brain Function
Your browsing activity is empty.
Activity recording is turned off.
See more...