Edema occurs when an excessive volume of fluid accumulates in the tissues, either within cells (cellular edema) or within the collagen-mucopolysaccharide matrix distributed in the interstitial spaces (interstitial edema) [14,42,62,64,87,88,141,215,247,279]. Our focus is on swelling of the extracellular matrix or interstitial edema, which may occur as a result of aberrant changes in the pressures (hydrostatic and oncotic) acting across the microvascular walls, alterations in the molecular structures that comprise the barrier to fluid and solute flux in the endothelial wall that are manifest as changes in hydraulic conductivity and the osmotic reflection coefficient for plasma proteins, or alterations in the lymphatic outflow system, as predicted by examination of the Starling equation.
Excessive accumulation of interstitial fluid is generally viewed as detrimental to tissue function because edema formation increases the diffusion distance for oxygen and other nutrients, which may compromise cellular metabolism in the swollen tissue. For the same reason, edema formation also limits the diffusional removal of potentially toxic byproducts of cellular metabolism. These are especially important problems in the lungs, where pulmonary edema can significantly impair gas exchange. In some tissues, certain anatomical structures limit the expansion of the tissue spaces in response to edemagenic stress. For example, the kidneys are enveloped by a tough fibrous capsule, the brain is surrounded by the cranial vault, and skeletal muscles in the volar and anterior tibial compartments are encased in tight fascial sheaths. As a consequence of the inability of these tissues to readily expand their interstitial volume, relatively small increments in transcapillary fluid filtration induce large increases in interstitial fluid pressure. This, in turn, reduces the vascular transmural pressure gradient and physically compresses capillaries, thereby reducing nutritive tissue perfusion [120]. In the intestine, unrestrained transcapillary filtration leads to exudation of interstitial fluid into the gut lumen, a phenomenon referred to as filtration-secretion or secretory filtration [87]. Filtration-secretion may compromise the absorptive function of the delicate intestinal mucosa and appears to occur as a result of the formation of large channels between mucosal cells in the villous tips when interstitial fluid pressure increases by greater than 5 mmHg [87]. Ascites, or the pathologic accumulation of fluid in the peritoneal cavity, occurs in cirrhosis and is caused by fluid weeping from congested hepatic sinusoids secondary to elevated portal venous pressure [223]. Ascites can predispose afflicted individuals to peritoneal infections, hepatic hydrothorax, and abdominal wall hernias [223].
Hydrostatic edema refers to accumulation of excess interstitial fluid which results from elevated capillary hydrostatic pressure while permeability edema results from disruption of the physical structure of the pores in the microvascular membrane such that the barrier is less able to restrict the movement of macromolecules from the blood to interstitium. Lymphedema represents a third form and may result from impaired lymph pump activity, an increase in lymphatic permeability favoring protein flux from lumen to interstitial fluid, lymphatic obstruction (e.g., microfiliarisis), or surgical removal of lymph nodes, as occurs in the treatment of breast cancer. Destruction of extracellular matrix proteins, as occurs in inflammation secondary to the formation of reactive oxygen and nitrogen species and release of hydrolytic enzymes from infiltrating leukocytes, resident immune cells, and cells comprising the tissue parenchyma, alters the compliance characteristics of interstitial gel matrix such that interstitial fluid pressure fails to increase and oppose the movement of fluid. In addition, the tensional forces that are normally exerted by extracellular matrix proteins on the anchoring filaments (Figure 3.1) attached to lymphatic endothelial cells to facilitate lymphatic filling are diminished as a result of disrupted mechanical integrity [249]. Reductions in circulating plasma proteins, especially albumin, produce edema by decreasing plasma colloid osmotic pressure, and occurs in liver disease and severe malnutrition.
4.1. The Margin of Safety Against Edema Formation – Edema Safety Factors
While increases in capillary pressure, reductions in plasma oncotic pressure, and/or disruption of endothelial barrier function are all accompanied by an increase in transmicrovascular filtration, the accumulation of fluid is resisted by a number of edema safety factors that work in concert to limit edema formation. This margin of safety against edema formation was first recognized in 1932 by Krogh and coworkers [148] as a means to explain why elevations in venous pressure by 10–15 mmHg failed to cause substantial accumulation of tissue fluid. Only when venous hypertension exceeded these levels did gross edema form, indicating that the margin of safety against edema formation could be overwhelmed. From the Starling equation (Equation (1.4)), one can readily see that increases in interstitial fluid pressure, reductions in tissue colloid osmotic pressure or microvascular surface area for exchange, or increases in lymph flow may all act to limit accumulation of excess fluid, and thus represent important edema safety factors against edema formation (Figures 4.1–4.5).
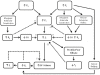
Figure 4.1
Mechanisms of enhanced transcapillary filtration in response to elevations in arterial or venous pressure. Elevations in arterial (Pa) or venous (Pv) pressure increase capillary pressure, which favors enhanced capillary filtration (Jv). The resulting (more...)
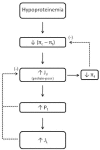
Figure 4.2
Hypoproteinemia reduces the effective colloid osmotic pressure gradient (πc − πt), resulting in increases in transcapillary fluid flux (JV). The resulting increase in interstitial fluid volume raises interstitial fluid pressure (more...)
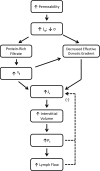
Figure 4.3
Increased microvascular permeability results in the formation of a protein-rich filtrate that raises interstitial colloid osmotic pressure (πt), thereby reducing the effective colloid osmotic pressure gradient (σ(πc − π (more...)
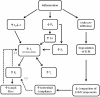
Figure 4.4
Inflammation results in the release of mediators that cause vasodilation, increase microvascular permeability, and induce leukocyte infiltration. Relaxation of vascular smooth muscle cells in arterioles and precapillary sphincters results in a reduction (more...)
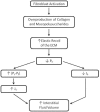
Figure 4.5
Myxedema is due to an accumulation of mucopolysaccharides secondary to overproduction of fibroblasts. This creates a suction force due to enhanced elastic recoil of the extracellular matrix that creates a high negative interstitial fluid pressure (Pt (more...)
In addition to these basic compensatory mechanisms, the myogenic response to increased wall tension in arterioles and venous bulging constitute other edema safety factors in response to elevations in arterial or venous pressure in some tissues (Figure 4.1) [88]. Myogenic arteriolar vasoconstriction attenuates the rise in capillary pressure that might otherwise occur in response to arterial or venous hypertension, and also acts to reduce the microvascular surface area available for fluid exchange secondary to precapillary sphincter closure [55,118,131,172]. When venous pressure is elevated, the volume of blood within postcapillary venules, larger venules and veins increases and bulge into the extravascular compartment, thereby raising tissue pressure. In effect, venous bulging stiffens the extracellular matrix by increasing tensional forces on the reticular fibers and fluid in this space [88]. Finally, changes in excluded volume with increased transcapillary fluid filtration also comprise an important component of the margin of safety against swelling of the extracellular matrix compartment [88,280].
From the aforementioned discussion, it is obvious that tissues exhibiting restrictive endothelial barrier properties, lowest interstitial compliance, and highest sensitivity of lymph flow to changes in interstitial fluid pressure will exhibit the greatest margin of safety against edema formation. Even in tissues where the endothelial barrier is less restrictive and lymphatic sensitivity is low, the margin of safety can still be quite substantial if the interstitial matrix is stiff.
4.2. Vasogenic Edema
Disturbances in the vascular compartment are among the most common causes of interstitial edema (vasogenic edema) and result from capillary hypertension or hypoproteinemia. Capillary pressure (Pc) is determined by arterial (PA) and venous (PV) pressure and the ratio of pre- to postcapillary resistances (RA/RV) as shown by the equation [197]:
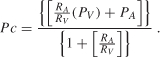
From Equation (4.1), it is apparent that capillary pressure rises when arterial or venous pressure increases and/or the pre- to postcapillary resistance ratio falls. Since arterial and venous pressure and the pre-to-postcapillary resistance ratio can be modified on a moment-to-moment basis in various physiologic (e.g., exercise) or pathologic conditions (e.g., inflammation) or following administration of vasoactive pharmaceutical agents, it might be expected that capillary pressure and thus transmicrovascular filtration rate can rapidly increase in accord with these changes. However, it has been suggested that capillary pressure may be tightly regulated in response to changes in arterial or venous pressure, by appropriate adjustments in pre- or postcapillary resistance, as a means to maintain a relatively constant interstitial fluid volume when any of these variables change [24,55,118,173]. For example, because vascular smooth muscle in arterial and arteriolar walls contracts when exposed to elevated intravascular pressures, this myogenic response increases precapillary resistance and protects capillaries from a concomitant rise in their intravascular pressure. Conversely, when arterial pressure falls, myogenic tone is reduced in arterioles, decreasing their resistance to flow and maintaining capillary pressure. These observations suggest that capillary pressure may be regulated over the same range of pressure changes over which flow is autoregulated in a given organ. Indeed, from the relation:
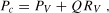
Similarly, changes in capillary pressure, and thus capillary filtration, are buffered when venous pressure is elevated [55,125,147]. At least two mechanisms account for this regulation of capillary pressure (Figure 4.1). Myogenic contraction of vascular smooth muscle in the walls of arterioles is elicited by transmission of the venous pressure increase to these upstream vessels [54,55,240]. A venous-arteriolar reflex has also been implicated in this response, wherein elevations in venous pressure activate antidromic impulses that are transmitted to nerve endings impinging on upstream arterioles, where neurotransmitter release elicits constriction [92,234]. However, more recent work has challenged the importance of this mechanism versus the myogenic response [206]. It is important to note that capillary pressure, and thus capillary filtration, is not as well regulated in response to increases in venous pressure or resistance as when arterial pressure is altered [55,144]. However, potential effects of increased venous pressure to reduce the capillary filtration coefficient may buffer the response to altered capillary pressure on transmicrovascular fluid movement, as outlined above.
While the aforementioned discussion focused on the effect of acute changes in venous pressure on the regulation of capillary pressure and transmicrovascular fluid movement and applies to most organs, the small intestinal vasculature may be unique in its response to chronic changes in venous pressure. Chronic intestinal venous hypertension induced by calibrated stenosis of the portal vein is associated with the development of a hyperdynamic circulation characterized by increased cardiac output, reduced intestinal vascular resistance, and increased intestinal blood flow [18,19,21,147]. The latter changes result in a larger increase in intestinal capillary pressure than occurs during acute venous pressure elevations of the same magnitude and are associated with increases in the capillary filtration coefficient [147]. As a consequence, the increase in transcapillary filtration is much greater in chronic versus acute venous hypertension. The mechanisms responsible for the reduction in intestinal vascular resistance that account for the changes in capillary pressure and capillary filtration coefficient that lead to enhanced capillary filtration in chronic portal hypertension involve the formation of vasodilator substances and other factors and are reviewed elsewhere [18,19,21,81,115,122,146].
Capillary pressure is only modestly increased (~2 mmHg) in chronic arterial hypertension because the increase in arterial resistance that causes the rise in arterial blood pressure buffers transmission of the pressure increase to the capillary level [145]. Nevertheless, the associated increase in transmicrovascular filtration rate largely accounts for the elevated transcapillary escape rate of proteins noted in this disorder through convective coupling of fluid and protein flux. Elevated capillary pressure and filtration rate occur early in the course of development of diabetes mellitus and is thought to be an important stimulus for capillary basement membrane thickening, the ultrastructural hallmark of diabetic microangiopathy [27,143]. Microvascular rarefaction, or loss of capillaries, has been reported to accompany the development of arterial hypertension, diabetes mellitus, and the metabolic syndrome [8,27,32,70,73,143]. The attendant reductions in the surface area available for exchange may partially offset the effect of capillary hypertension to increase interstitial fluid volume in these conditions.
Very large increases in venous pressure may induce increments in capillary filtration far in excess of what would be predicted from the associated increase in capillary pressure. This is due to pressure-induced increases in microvascular permeability that are manifest in the Starling equation by increases in hydraulic conductivity and reductions in the osmotic reflection coefficient. For most organs, the permeability characteristics of the microvascular barrier to the exchange of fluid and lipid-insoluble solutes can be explained by the existence of large numbers of small pores with radii of 70 angstroms or less and a smaller number of large pores with radii in excess of 200 angstroms, with some models incorporating a third set of very small pores (< 10 angstroms in radius) to account for the diffusional flux of water. (Organs such as the liver, which have discontinuous capillaries characterized by large gaps between endothelial cells and reflection coefficients approaching 0.1, do not fit these models). Large increases in venous pressure are thought to enlarge these pores in microvascular wall, which is referred to as the stretched pore phenomenon [199,218,238]. Individual organs demonstrate a differential sensitivity to the effect of elevated venous pressure with regard to induction of the stretch pore phenomenon. For example, no increase in permeability occurs in microvessels of the feet during quiet standing, even though capillary pressure in the feet increases by more than 50 mmHg relative to values measured when supine, owing to the large hydrostatic column in arteries and veins. However, pulmonary capillaries may demonstrate a stretched pore phenomenon during conditions such as left ventricular failure, an effect that exacerbates pulmonary edema formation in this condition [199].
As noted above, myogenic constriction of arterioles in response to elevations in arterial or venous pressure constitutes an important safety factor against edema formation in hydrostatic edema by limiting the increase in capillary pressure and by reducing the number of perfused capillaries, and thus the available surface area for fluid filtration, that might otherwise occur in response to arterial or venous hypertension or increased venous resistance (Figure 4.1). However, it is important to note even modest increments in capillary pressure, which might appear to be small and inconsequential, can result in substantial increases in fluid filtration rates across the microvasculature. This is because normal net filtration pressure is quite small, averaging 0.15 mmHg for a prototypical body capillary. Thus, increasing capillary pressure by just 2 mmHg, as noted above in arterial hypertension, results in an initial 14-fold increase in fluid movement from the blood into the interstitium. Capillary hypertension results in the formation of a protein-poor ultrafiltrate that upon entry into the interstitial space raises interstitial fluid volume. Owing to the compliance characteristics of the interstitium, small increments in interstitial volume produce very large increases in tissue pressure, which effectively reduces the transcapillary hydrostatic pressure gradient, thereby limiting further accumulation of fluid (Figure 4.1). This effect is exacerbated in response to elevations in venous outflow pressure through the phenomenon of venous bulging. That is, the volume in veins increases immediately on elevation of venous pressure, which produces a coincident increase in interstitial pressure caused by expansion of engorged venules and veins into the interstitial spaces (Figure 4.1). In essence, venous engorgement shifts the interstitial compliance curve to the left, so that a smaller change in interstitial volume produces a larger increase in interstitial pressure. Increased interstitial fluid pressure increases lymph flow by three mechanisms. First, increased tissue pressure provides the driving pressure for flow into initial lymphatics. Second, increased pressure in the interstitial compartment creates radial tension on the anchoring filaments connecting the extracellular matrix to lymphatic endothelial cells, locally increasing initial lymphatic diameter and opening gaps between interdigitating and overlapping junctions between adjacent lymphatic endothelial cells (Figure 3.1). These tensional forces create a small, transient suction pressure for movement of interstitial fluid through enlarged gaps between adjacent endothelial cells, which act as a second, one-way valve system to ensure unidirectional flow from the interstitium into lymphatics. Third, as fluid moves into initial lymphatics, it increases volume in upstream lymphangions, promoting their contractile activity and lymph flow. The presence of valves between adjacent lymphangions assures one-way flow.
As noted above, capillary hypertension results in the movement of protein-poor fluid into the interstitial spaces, reducing the concentration of tissue proteins and decreasing tissue colloid osmotic pressure (Figure 4.1). This increases the effectiveness of the transcapillary oncotic pressure gradient (πc − πt) in opposing the hydrostatic gradient (Pc − Pt) favoring filtration. Because solute is excluded from a large portion of gel water in the extracellular matrix, the rapidity of the decrease in tissue protein concentration that occurs in response to increased interstitial fluid volume is enhanced, thereby augmenting the effectiveness of protein washdown as an edema safety factor. It is important to note that the effectiveness of decreases in tissue osmotic pressure as an edema safety factor is reduced in severe capillary hypertension, owing to the stretched-pore phenomenon discussed above, which increases convective-coupled protein transport into the tissue spaces.
4.3. Hypoproteinemia
Marked reductions in the circulating levels of proteins, especially albumin, is another cause of edema that relates to intravascular factors (Figure 4.2). Hypoproteinemia may result from rapid loss of proteins across a compromised glomerular barrier in diseased kidneys, impaired hepatic synthesis of plasma proteins in liver disease, severe malnutrition or protein-losing enteropathy (which limits the availability of substrate for protein synthesis), or from infusion of intravenous fluids lacking macromolecules. The ensuing reduction in the colloid osmotic pressure gradient (πc − πt), which favors reabsorption in the non-steady state and opposes the hydrostatic pressure gradient that favors filtration, induced by hypoproteinemia can result in a large transcapillary flux of protein-poor fluid into the interstitial spaces (Figure 4.2). Like capillary hypertension, this effect is opposed by elevations in tissue hydrostatic pressure, which increases lymph flow, both of which serve to limit the accumulation of tissue fluid (Figure 4.2). Enhanced capillary filtration also acts to dilute the concentration of proteins in the extracellular spaces, an effect that is magnified by increasing the accessible volume in the extracellular matrix gel (Figures 2.1 and 4.2). The ensuing reduction in interstitial colloid osmotic pressure acts to reduce net filtration pressure, thereby minimizing edema formation. Unlike the response to vascular hypertension, there is no stimulus for myogenic arteriolar vasoconstriction and venous bulging does not occur in hypoproteinemia, which reduces the margin of safety for edema formation in response to this edemagenic stress. As a consequence, tissues are less able to compensate for reductions in plasma colloid osmotic pressure that are equivalent to a given increase in capillary hydrostatic pressure.
4.4. Permeability Edema and Inflammation
Disruption of the microvascular barrier is a pathologic sequela in a large number of disease states, commonly accompanies trauma, and can be induced by a wide variety of endogenously produced mediators and pharmacologic agents. In the Starling equation (Equation (1.4)), this increase in permeability is manifest as a reduction in the osmotic reflection coefficient and/or an increase in hydraulic conductivity (Figure 4.3). Rapid reductions in the reflection coefficient decrease the effectiveness of the colloid osmotic pressure gradient in opposing filtration. The reduction in the restrictive properties of the endothelial barrier allows movement of a protein-rich filtrate into the tissue spaces, which increases interstitial colloid osmotic pressure (Figure 4.3). The resultant reduction in the colloid osmotic pressure gradient increases net filtration pressure, an effect that is exacerbated by the fact that many if not most of the mediators that increase microvascular permeability also act as vasodilators and reduce arteriolar resistance (Figure 4.4). As a consequence, capillary pressure is elevated, which further increases net filtration pressure. In addition, vasodilatation tends to recruit capillaries, thereby increasing microvascular surface area available for fluid and protein flux into the tissues. The latter change contributes to a further increase in the capillary filtration coefficient (which is equal to the hydraulic conductivity times surface area, LpS), thereby magnifying the effect of increased net filtration pressure to promote volume flux. The marked enhancement in transcapillary fluid filtration results in increased convective transport of protein through the enlarged pores in the microvascular barrier (Figure 4.3). Under such conditions, the effect of increases in interstitial fluid pressure and lymph flow to provide a margin of safety against edema formation are rapidly overwhelmed and marked swelling of the interstitial spaces ensues.
Permeability edema is exacerbated in inflammatory states that are characterized by leukocyte infiltration into the tissues (Figure 4.4). Inflammation is a characteristic response to tissue injury and involves the release of a large number of mediators that not only increase microvessel permeability and cause vasodilatation, but also act to attract leukocytes to the damaged tissue (Figure 4.4). These phagocytic cells release a variety of hydrolytic enzymes as well as reactive oxygen and nitrogen species that degrade extracellular matrix components and the anchoring filaments that attach to lymphatic endothelial cells (Figures 3.1 and 4.4). This reduces the radial tension on the valve-like overlapping and interdigitating cell membranes at the interendothelial junctions in initial lymphatics, which may compromise lymphatic filling. Leukocyte-mediated disruption of the extracellular matrix components also increases interstitial compliance, which allows a larger volume of extracellular fluid to be accommodated within the matrix with little increase in interstitial fluid pressure, thereby attenuating the effectiveness of this edema safety factor. This effect is exacerbated by disruption of the connections fibroblasts form with collagen fibers in the interstitial spaces, which normally help compact the extracellular matrix by imposing tensional forces on these fibrillar components and restrain the gel matrix from taking up fluid and swelling (Figure 2.3). Extracellular matrix disruption thus produces a more compliant interstitium that limits the increase in interstitial fluid pressure for a given change in interstitial volume. Excluded volumes are also reduced by matrix degradation, an effect that increases effective interstitial colloid osmotic pressure. Extravasated proteins move more readily through the disrupted matrix, facilitating blood-to-lymph transport of these macromolecules.
4.5. Neurogenic Inflammation
Neurogenic inflammation is characterized by leukosequestration, edema formation, and extravasation of plasma proteins following stimulation of sensory neurons. Sensory fibers release calcitonin gene-related peptide, substance P, and neurokinin A when stimulated. These proinflammatory molecules may act directly on the microvasculature to produce inflammation, but also appear to activate tissue mast cells, which augment the inflammatory response by release of their own complement of mediators.
4.6. Myxedema
Myxedema is caused by suction of plasma filtrate into the tissue spaces that occurs as a result of overproduction of interstitial collagen and mucopolysaccharides by fibroblasts. Increasing the density of these extracellular matrix components augments the elastic recoil of the interstitial gel matrix and thereby producing a highly negative interstitial fluid pressure (Figure 4.5). As a consequence, lymph flow is reduced. Increased matrix density also increases the excluded volume, which acts to increase the effective interstitial colloid osmotic pressure. In effect, these changes create a suction force that accelerates fluid filtration and the development of edema. The most frequent manifestation of myxedema occurs in cases of hypothyroidism secondary to increased deposition of tissue matrix [149].
4.7. Lymphedema
Because lymphatic drainage represents the major route for removal of interstitial fluid (and macromolecules) formed by capillary filtration, dysfunction of lymphatic vessels causes the development of edema and can exacerbate edema induced by other causes (Figure 4.6). Lymphedema occurs with physical obstruction of the lymphatic vessel lumen (either by extramural forces exerted by tumors or intraluminal obstruction by metastasizing tumor cells), destruction or regression of existing lymphatics, incompetence of the valves between lymphangions, paralysis of lymphatic muscle, reduced tissue motion, diminished arterial pulsations or venomotion, or by elevated venous pressure at the drainage points where lymphatics empty into the systemic blood circulation (Figure 4.6). Whatever the cause of lymphatic dysfunction, edema formation does not occur until lymph flow is reduced by 50%, all other factors being equal.
Figure 4.6
Lymphedema arises in response to a variety of conditions that result in reduced lymph flow. When lymphatic outflow (JL) is completely occluded, interstitial fluid volume initially increases because capillary filtration (JV) occurs until the interstitial (more...)
In the case of complete obstruction, lymph flow draining a tissue region falls to zero. Transcapillary filtration into this tissue region continues until interstitial pressure rises to equal net filtration pressure. As transcapillary volume flux decreases, the convective transport of protein from the vascular to interstitial compartments decreases. Since extravasated protein is not removed by the obstructed lymphatic, diffusional flux of protein continues until the concentration gradient is dissipated. At equilibrium, interstitial fluid pressure rises to microvascular hydrostatic pressure and interstitial colloid osmotic pressure equals plasma colloid osmotic pressure, yielding a net filtration pressure of zero. The affected tissue is characterized by large increases in water and protein content, fibrosis, and adipose cell deposition.
Publication Details
Copyright
Publisher
Morgan & Claypool Life Sciences, San Rafael (CA)
NLM Citation
Scallan J, Huxley VH, Korthuis RJ. Capillary Fluid Exchange: Regulation, Functions, and Pathology. San Rafael (CA): Morgan & Claypool Life Sciences; 2010. Chapter 4, Pathophysiology of Edema Formation.

%2C%20resulting%20in%20increases%20in%20transcapillary%20fluid%20flux%20(JV).&p=BOOKS&id=53445_fig4.2.jpg "Click on image to zoom")
%2C%20thereby%20reducing%20the%20effective%20colloid%20osmotic%20pressure%20gradient%20(%003C3(%003C0c%20%02212%20%003C0t))%20acting%20across%20the%20microvascular%20wall.&p=BOOKS&id=53445_fig4.3.jpg "Click on image to zoom")


