By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2000, Geoffrey M Cooper.
Bookshelf ID: NBK9950

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000.

Classical experiments in molecular biology were strikingly successful in developing our fundamental concepts of the nature and expression of genes. Since these studies were based primarily on genetic analysis, their success depended largely on the choice of simple, rapidly replicating organisms (such as bacteria and viruses) as models. It was not clear, however, how these fundamental principles could be extended to provide a molecular understanding of the complexities of eukaryotic cells, since the genomes of most eukaryotes (e.g., the human genome) are up to a thousand times more complex than that of E. coli. In the early 1970s, the possibility of studying such genomes at the molecular level seemed daunting. In particular, there appeared to be no way in which individual genes could be isolated and studied.
This obstacle to the progress of molecular biology was overcome by the development of recombinant DNA technology, which provided scientists with the ability to isolate, sequence, and manipulate individual genes derived from any type of cell. The application of recombinant DNA has thus enabled detailed molecular studies of the structure and function of eukaryotic genes, thereby revolutionizing our understanding of cell biology.
The first step in the development of recombinant DNA technology was the characterization of restriction endonucleases—enzymes that cleave DNA at specific sequences. These enzymes were identified in bacteria, where they apparently provide a defense against the entry of foreign DNA (e.g., from a virus) into the cell. Bacteria have a variety of restriction endonucleases that cleave DNA at more than a hundred distinct recognition sites, each of which consists of a specific sequence of four to eight base pairs (examples are given in Table 3.2).
Recognition Sites of Representative Restriction Endonucleases.
Since restriction endonucleases digest DNA at specific sequences, they can be used to cleave a DNA molecule at unique sites. For example, the restriction endonuclease EcoRI recognizes the six-base-pair sequence GAATTC. This sequence is present at five sites in DNA of the bacteriophage λ, so EcoRI digests λ DNA into six fragments ranging from 3.6 to 21.2 kilobases long (1 kilobase, or kb = 1000 base pairs) (Figure 3.16). These fragments can be separated according to size by gel electrophoresis—a common method in which molecules are separated based on the rates of their migration in an electric field. A gel, usually formed from agarose or polyacrylamide, is placed between two buffer compartments containing electrodes. The sample (e.g., the mixture of DNA fragments to be analyzed) is then pipetted into preformed slots in the gel, and the electric field is turned on. Nucleic acids are negatively charged (because of their phosphate backbone), so they migrate toward the positive electrode. The gel acts like a sieve, selectively retarding the movement of larger molecules. Smaller molecules therefore move through the gel more rapidly, allowing a mixture of nucleic acids to be separated on the basis of size.
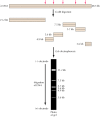
EcoRI digestion and gel electrophoresis of λ DNA. EcoRI cleaves λ DNA at five sites (arrows), yielding six DNA fragments. These fragments are then separated by electrophoresis in an agarose gel. The DNA fragments migrate toward the positive (more...)
In addition to size, the order of restriction fragments can be determined by a variety of methods, yielding (for example) a map of the EcoRI sites in λ DNA. The locations of cleavage sites for multiple different restriction endonucleases can be used to generate detailed restriction maps of DNA molecules, such as viral genomes (Figure 3.17). In addition, individual DNA fragments produced by restriction endonuclease digestion can be isolated following electrophoresis for further study—including determination of their DNA sequence. The DNAs of many viruses have been characterized by this approach.
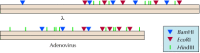
Restriction maps of λ and adenovirus DNAs. The locations of cleavage sites for BamHI, EcoRI, and HindIII are shown in the DNAs of E. coli bacteriophage λ (48.5 kb) and human adenovirus-2 (35.9 kb).
Restriction endonuclease digestion alone, however, does not provide sufficient resolution for the analysis of larger DNA molecules, such as cellular genomes. A restriction endonuclease with a six-base-pair recognition site (such as EcoRI) cleaves DNA with a statistical frequency of once every 4096 base pairs (1/46). A molecule the size of λ DNA (48.5 kb) would therefore be expected to yield about ten EcoRI fragments, consistent with the results illustrated in Figure 3.16. However, restriction endonuclease digestion of larger genomes yields quite different results. For example, the human genome is approximately 3 × 106 kb long and is therefore expected to yield more than 500,000 EcoRI fragments. Such a large number of fragments cannot be separated from one another, so agarose gel electrophoresis of EcoRI-digested human DNA yields a continuous smear rather than a discrete pattern of DNA fragments. Because it is impossible to isolate single restriction fragments from such digests, restriction endonuclease digestion alone does not yield a source of homogeneous DNA suitable for further analysis. Quantities of such purified DNA fragments, however, can be obtained through molecular cloning.
The basic strategy in molecular cloning is to insert a DNA fragment of interest (e.g., a segment of human DNA) into a DNA molecule (called a vector) that is capable of independent replication in a host cell. The result is a recombinant molecule or molecular clone, composed of the DNA insert linked to vector DNA sequences. Large quantities of the inserted DNA can be obtained if the recombinant molecule is allowed to replicate in an appropriate host. For example, fragments of human DNA can be cloned in bacteriophage λ vectors (Figure 3.18). These recombinant molecules can then be introduced into E. coli, where they replicate efficiently to yield millions of progeny phages containing the human DNA insert. The DNA of these phages can then be isolated, yielding large quantities of recombinant molecules containing a single fragment of human DNA. Whereas this fragment might represent one part in 100,000 of human genomic DNA, it represents approximately one part in 10 after being cloned in the λ vector. Moreover, the fragment can be easily isolated from the rest of the vector DNA by restriction endonuclease digestion and gel electrophoresis, allowing a pure fragment of human DNA to be analyzed and further manipulated.
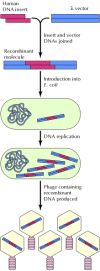
Generation of a recombinant DNA molecule. A fragment of human DNA is inserted into a λ DNA vector. The resulting recombinant molecule is then introduced into E. coli, where it replicates to yield recombinant progeny phage containing the human (more...)
The DNA fragments used to create recombinant molecules are usually generated by digestion with restriction endonucleases. Many of these enzymes cleave their recognition sequences at staggered sites, leaving overhanging or cohesive single-stranded tails that can associate with each other by complementary base pairing (Figure 3.19). The association between such paired complementary ends can be established permanently by treatment with DNA ligase, an enzyme that seals breaks in DNA strands (see Chapter 5). Thus, two different fragments of DNA (e.g., a human DNA insert and a λ DNA vector) prepared by digestion with the same restriction endonuclease can be readily joined to create a recombinant DNA molecule.
Joining of DNA molecules. Vector and insert DNAs are digested with a restriction endonuclease (such as EcoRI), which cleaves at staggered sites leaving overhanging single-stranded tails. Vector and insert DNAs can then associate by complementary base (more...)
The fragments of DNA that can be cloned are not limited to those that terminate in restriction endonuclease cleavage sites. Synthetic DNA “linkers” containing a variety of restriction endonuclease sites can be added to the blunt ends of any DNA fragment. Linkers are short oligonucleotides that can be readily obtained by chemical synthesis, allowing virtually any fragment of DNA to be prepared for ligation to a vector.
Not only DNA, but also RNA sequences can be cloned (Figure 3.20). The first step is to synthesize a DNA copy of the RNA using the enzyme reverse transcriptase. The DNA product (called a cDNA because it is complementary to the RNA used as a template) can then be ligated to vector DNA as already described. Since eukaryotic genes are usually interrupted by noncoding sequences (introns; see Chapter 4), which are removed from mRNA by splicing, the ability to clone cDNA as well as genomic DNA has been critical for understanding gene structure and function.
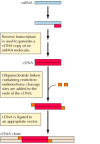
cDNA cloning.
Depending on the size of the insert DNA and the purpose of the experiment, many different types of cloning vectors can be used for the generation of recombinant molecules. The basic vector systems used for the isolation and propagation of cloned DNAs are reviewed here. Other vectors developed for the expression of cloned DNAs and the introduction of recombinant molecules into eukaryotic cells are discussed in subsequent sections.
Bacteriophage λ vectors are frequently used for the initial isolation of either genomic or cDNA clones from eukaryotic cells (Figure 3.21). In λ cloning vectors, sequences of the bacteriophage genome that are dispensable for virus replication have been removed and replaced with unique restriction sites for insertion of cloned DNA. DNA inserts can be as large as about 15 kb and still yield a recombinant genome that can be packaged into phage particles. To isolate genomic clones of human DNA, for example, random fragments of human DNA with an average size of about 15 kb are ligated to λ vector arms. These recombinant DNA molecules can then be efficiently packaged into phage particles by mixing DNA with λ proteins (called packaging extracts) in vitro. The phage particles are then used to infect cultures of E. coli. Since each recombinant phage forms a single plaque, recombinants carrying unique inserts of human DNA can be isolated. In addition, recombinant phages containing particular genes of interest can be identified by nucleic acid hybridization or other screening methods, as discussed in the next section.

Cloning in bacteriophage λ vectors. The vector contains a restriction site (e.g., an EcoRI site) for insertion of cloned DNA. In addition, cos sites (cohesive ends), which are required for packaging DNA into phage particles, are present on both (more...)
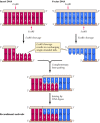
Plasmid vectors (Figure 3.22) allow easier manipulation of cloned DNA sequences than do phage vectors. Plasmids are small circular DNA molecules that can replicate independently—without being associated with chromosomal DNA—in bacteria. All that is required on the plasmid DNA is an origin of replication—the DNA sequence that signals the host cell DNA polymerase to replicate the DNA molecule. In addition, plasmid vectors carry genes that confer resistance to antibiotics (e.g., ampicillin resistance), so bacteria carrying the plasmids can be selected. Plasmid vectors usually consist of only 2 to 4 kb of DNA, in contrast to the 30 to 45 kb of phage DNA present in λ vectors, facilitating the analysis of an inserted DNA fragment.
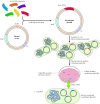
Cloning in plasmid vectors. The vector is a small circular molecule that contains an origin of replication (ori), a gene conferring resistance to ampicillin (Ampr), and a restriction site (e.g., EcoRI) which can be used to insert foreign DNA. Insert DNA (more...)
To be cloned into a plasmid vector, a fragment of the insert DNA is ligated to an appropriate restriction site in the vector and the recombinant molecule is used to transform E. coli. Antibiotic-resistant colonies, which contain plasmid DNA, are selected. Such plasmid-containing bacteria can then be grown in large quantities and their DNA extracted. The small circular plasmid DNA molecules, of which there are often hundreds of copies per cell, can be separated from the bacterial chromosomal DNA; the result is purified plasmid DNA that is suitable for analysis of the cloned insert.
For some studies involving analysis of genomic DNA, it is desirable to clone even larger fragments of DNA than are accommodated by λ vectors. Cosmid and yeast artificial chromosome (YAC) vectors can be used for this purpose. Cosmid vectors (Figure 3.23) accommodate inserts of approximately 45 kb. These vectors contain bacteriophage λ sequences that allow efficient packaging of the cloned DNA into phage particles. In addition, cosmids contain origins of replication and the genes for antibiotic resistance that are characteristic of plasmids, so they are able to replicate as plasmids in bacterial cells. Even larger fragments of DNA (hundreds of kilobases) can be cloned in YAC vectors, which replicate as chromosomes in yeast cells. These vectors are particularly useful for chromosome mapping studies, as discussed in Chapter 4.
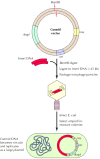
Cloning in cosmid vectors. A cosmid is a plasmid containing cos sites, which allow DNA to be packaged into λ phage particles. Large fragments of insert DNA (approximately 45 kb) are ligated to a cloning site (e.g., BamHI) to yield molecules of (more...)
Molecular cloning allows the isolation of individual fragments of DNA in quantities suitable for detailed characterization, including the determination of nucleotide sequence. Indeed, determination of the nucleotide sequences of many genes has elucidated not only the structure of their protein products, but also the properties of DNA sequences that regulate gene expression. Furthermore, the coding sequences of novel genes are frequently related to those of previously studied genes, and the functions of newly isolated genes can often be correctly deduced on the basis of such sequence similarities.
Current methods of DNA sequencing are both rapid and accurate, and determining the sequence of several kilobases of DNA is a straightforward task for most molecular biology laboratories. Thus, it is now far easier to clone and sequence DNA than it is to determine the amino acid sequence of a protein. Since the nucleotide sequence of a gene can be readily translated into the amino acid sequence of its encoded protein, the easiest way of determining protein sequence is the sequencing of a cloned gene.
The most common method of DNA sequencing is based on premature termination of DNA synthesis resulting from the inclusion of chain-terminating dideoxynucleotides (which do not contain the deoxyribose 3′ hydroxyl group) in DNA polymerase reactions (Figure 3.24). DNA synthesis is initiated from a primer that has been labeled at one end with a radioisotope. Four separate reactions are run, each including one dideoxynucleotide (either A, C, G, or T) in addition to its normal counterpart. Incorporation of a dideoxynucleotide stops further DNA synthesis because no 3′ hydroxyl group is available for addition of the next nucleotide. Thus, a series of labeled DNA molecules is generated, each terminating at the base represented by the dideoxynucleotide in each reaction. These fragments of DNA are then separated according to size by gel electrophoresis and detected by exposure of the gel to X-ray film (autoradiography). The size of each fragment is determined by its terminal dideoxynucleotide, so the DNA sequence corresponds to the order of fragments read from the gel.

DNA sequencing by the Sanger procedure. Dideoxynucleotides, which lack OH groups at the 3′ as well as the 2′ position of deoxyribose, are used to terminate DNA synthesis at specific bases. These molecules are incorporated normally into (more...)
Large-scale DNA sequencing is frequently performed using automated systems, which use fluorescence-labeled primers in dideoxynucleotide sequencing reactions (Figure 3.25). As the newly syntehsized DNA strands are electrophoresed through a gel, they pass through a laser beam that excites the fluorescent label. The resulting emitted light is then detected by a photomultiplier, and a computer collects and analyzes the data. This type of automated DNA sequencing has enabled the large-scale analysis required for determination of the complete genome sequences of bacteria, yeast, C. elegans, and Drosophila, and is soon expected to yield the complete sequence of the human genome.
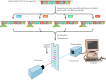
Automated DNA sequencing. Four separate sequencing reactions are performed, each containing one chain-terminating dideoxynucleotide and a primer labeled with a distinct fluorescent tag. The products are then pooled and subjected to gel electrophoresis. (more...)
In addition to enabling determination of the nucleotide sequences of genes—and hence the amino acid sequences of their protein products—molecular cloning has provided new approaches to obtaining large amounts of proteins for structural and functional characterization. Many proteins of interest are present at only low levels in eukaryotic cells and therefore cannot be purified in significant amounts by conventional biochemical techniques. Given a cloned gene, however, this problem can be solved by the engineering of vectors that lead to high levels of gene expression in either bacteria or eukaryotic cells.
To express a eukaryotic gene in E. coli, the cDNA of interest is cloned into a plasmid or phage vector (called an expression vector) that contains sequences that drive transcription and translation of the inserted gene in bacterial cells (Figure 3.26). Inserted genes often can be expressed at levels high enough that the protein encoded by the cloned gene corresponds to as much as 10% of the total bacterial protein. Purifying the protein encoded by the cloned gene in quantities suitable for detailed biochemical or structural studies is then a straightforward matter.
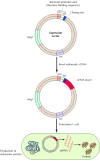
Expression of cloned genes in bacteria. Expression vectors contain promoter sequences (pro) that direct transcription of inserted DNA in bacteria and sequences required for binding of mRNA to bacterial ribosomes (Shine-Delgarno [SD] sequences). A eukaryotic (more...)
It is frequently useful to express high levels of a cloned gene in eukaryotic cells, rather than in bacteria. This mode of expression may be important, for example, to ensure that posttranslational modifications of the protein (such as addition of carbohydrates or lipids) occur normally. Such protein expression in eukaryotic cells can be achieved, as in E. coli, by insertion of the cloned gene into a vector (usually derived from a virus) that directs high-level gene expression. One system frequently used for protein expression in eukaryotic cells is infection of insect cells by baculovirus vectors, which direct very high levels of expression of genes inserted in place of a viral structural protein. Alternatively, high levels of protein expression can be achieved using appropriate vectors in mammalian cells. Expression of cloned genes in yeast is particularly useful because simple methods of yeast genetics can be employed to identify proteins that interact with other cloned proteins or with specific DNA sequences.
Molecular cloning allows individual DNA fragments to be propagated in bacteria and isolated in large amounts. An alternative method to isolating large amounts of a single DNA molecule is the polymerase chain reaction (PCR), which was developed by Kary Mullis in 1988. Provided that some sequence of the DNA molecule is known, PCR can achieve a striking amplification of DNA via reactions carried out entirely in vitro. Essentially, DNA polymerase is used for repeated replication of a defined segment of DNA. The number of DNA molecules increases exponentially, doubling with each round of replication, so a substantial quantity of DNA can be obtained from a small number of initial template copies. For example, a single DNA molecule amplified through 30 cycles of replication would theoretically yield 230 (approximately 1 billion) progeny molecules. Single DNA molecules can thus be amplified to yield readily detectable quantities of DNA that can be isolated by molecular cloning or further analyzed directly by restriction endonuclease digestion or nucleotide sequencing.
The general procedure for PCR amplification of DNA is illustrated in Figure 3.27. The starting material can be either a cloned DNA fragment or a mixture of DNA molecules—for example, total DNA from human cells. A specific region of DNA can be amplified from such a mixture, provided that the nucleotide sequence surrounding the region is known so that primers can be designed to initiate DNA synthesis at the desired point. Such primers are usually chemically synthesized oligonucleotides containing 15 to 20 bases of DNA. Two primers are used to initiate DNA synthesis in opposite directions from complementary DNA strands. The reaction is started by heating the template DNA to a high temperature (e.g., 95°C) so that the two strands separate. The temperature is then lowered to allow the primers to pair with their complementary sequences on the template strands. DNA polymerase then uses the primers to synthesize a new strand complementary to each template. Thus in one cycle of amplification, two new DNA molecules are synthesized from one template molecule. The process can be repeated multiple times, with a twofold increase in DNA molecules resulting from each round of replication.
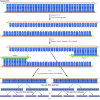
Amplification of DNA by PCR. The region of DNA to be amplified is flanked by two sequences used to prime DNA synthesis. The starting double-stranded DNA is heated to separate the strands and then cooled to allow primers (usually oligonucleotides of 15 (more...)
The multiple cycles of heating and cooling involved in PCR are performed by programmable heating blocks called thermocyclers. The DNA polymerases used in these reactions are heat-stable enzymes from bacteria such as Thermus aquaticus, which lives in hot springs at temperatures of about 75°C. These polymerases are stable even at the high temperatures used to separate the strands of double-stranded DNA, so PCR amplification can be performed rapidly and automatically. RNA sequences can also be amplified by this method if reverse transcriptase is used to synthesize a cDNA copy prior to PCR amplification.
If enough of the sequence of a gene is known that primers can be specified, PCR amplification provides an extremely powerful method of obtaining readily detectable and manipulable amounts of DNA from starting material that may contain only a few molecules of the desired DNA sequence in a complex mixture of other molecules. For example, defined DNA sequences of up to several kilobases can be readily amplified from total genomic DNA, or a single cDNA can be amplified from total cell RNA. These amplified DNA segments can then be further manipulated or analyzed, for example, to detect mutations within a gene of interest. PCR is thus a powerful addition to the repertoire of recombinant DNA techniques. Its power is particularly apparent in applications such as the diagnosis of inherited diseases, studies of gene expression during development, and forensic medicine.
Molecular Medicine: HIV and AIDS.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...