

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Extensive studies of E. coli glutaminyl-tRNA synthetase (GlnRS) over a greater than 30-year period have established this enzyme as an important paradigm for the class I tRNA synthetases. The later work in particular is distinguished by the interplay of genetic and biochemical experiments with atomic-resolution structures determined by X-ray crystallography. GlnRS has been a lead system for the application of genetic selections, for the delineation of detailed tRNA-enzyme contacts at atomic resolution, for the elucidation of the structural mechanism of catalysis, and for discovery of the interdependence of amino acid and tRNA specificities. Most recently, new approaches in aminoacylation kinetics and Xray crystallography open the possibility of moving towards a considerably more rigorous integration of structure and function. Together with insights derived from the increasing availability of newly sequenced glnS genes, this work may ultimately allow for successful re-engineering of the amino acid and tRNA specificities of the enzyme.
Introduction
Glutaminyl-tRNA synthetase (GlnRS) is the enzyme responsible for catalyzing the transfer of glutamine to the A76 2' hydroxyl group of tRNAGln isoacceptors. It has been extensively studied in many laboratories for over 30 years. A comprehensive review of the literature on GlnRS, comprising almost 200 publications, was published approximately five years ago.1 Therefore, this chapter will be complete in citations to the literature since that date, and selective in reference to the earlier work. The emphasis here will be on description and analysis of the most important structure-function characteristics of the E. coli enzyme, with a view towards distinguishing the important unresolved questions which still remain. Information on the purification of GlnRS enzymes from natural sources and on the regulation of glnS gene expression may be found in the earlier review.1 A shorter review of the structure-function relationships has been published as well.2 Reviews on tRNA recognition by GlnRS3-5 and on the early structural results6 have also appeared.
The dual specificity manifested by GlnRS is extraordinary yet typical of all tRNA synthetases, since strong discrimination against both noncognate amino acids and noncognate tRNAs is required to maintain the fidelity of protein synthesis. Crystal structures of the enzyme are available bound to both the cognate glutamine amino acid substrate and to tRNAGln.7-10 However, while these structures reveal the nature of the enzyme-substrate contacts in high-resolution detail, this is not sufficient to understand specificity, for two reasons. First, GlnRS discriminates among tRNAs using “indirect” as well as direct readout,11,12 so that the basis for selectivity is not apparent solely from examining contacts with base-specific functional groups. Second, it appears that the amino acid and tRNA specificities are coupled through the enzyme structure,13,14 with key signals likely arising from conformational changes which occur during formation of the specific complex. A consequence of these findings is that, while the identity of tRNAGln can be readily altered by replacements of relatively small numbers of nucleotides, neither the tRNA nor the amino acid specificities of GlnRS have been successfully re-engineered. It is clear that the outstanding question of how GlnRS distinguishes among globally similar tRNA and amino acid architectures remains elusive in spite of the available structural and biochemical data. An important purpose for this review is to explore the limitations in our understanding, and to suggest fruitful avenues for further progress.
Crystal Structures of GlnRS
Overview of Available Structures
E. coli GlnRS was first cocrystallized in the presence of tRNA2Gln and ATP in a centered orthorhombic lattice from solutions containing sodium citrate as precipitating agent.15 A structure of the ternary complex was determined at 2.8 Å resolution from these crystals.7,16 Identical crystals could also be grown from solutions in which the citrate was replaced by ammonium sulfate.7 The tRNA in this complex was obtained by in vivo overexpression.15 Subsequently, a crystal structure was determined using tRNA derived from in vitro transcription,17 which showed that the in vivo-derived tRNA in the original structures is at least partially unmethylated at the positions of the modified bases 2'-mG17, 2'-mU32, 2'-mA37, and T54. In vitro transcripts were later used to determine cocrystal structures of mutated tRNAGln species possessing altered tertiary core domains.18,19 Cocrystallization of the GlnRS-tRNAGln complex with the aminoacyl adenylate analog 5'-O-[N-(L-glutaminyl) sulfamoyl]adenosine (QSI) yielded a structure delineating the interactions made by glutamine in the amino acid binding pocket.10 Three cocrystal structures of GlnRS mutants, isolated by genetic selections based on mischarging phenotypes,16 have also been solved in the same crystal lattice.20
Very recently, two new approaches to the determination of GlnRS crystal structures have been developed. GlnRS was crystallized in a quaternary complex bound to tRNAGln, glutamine, and the ATP analog AMPCPP, under conditions similar to those of the ternary complexes bound to ATP and to QSI.21,120 AMPCPP does not support catalysis because the reactive α-phosphate is replaced by a methylene group. This provides a methodology suitable for determining structures bound to free amino acid substrates. Second, suitable crystals of unliganded GlnRS have been grown in a different orthorhombic lattice, and the structure of the enzyme in this state determined and refined to 2.65 Å resolution.121 Comparison of the unliganded and tRNA-bound enzyme yields detailed insight into the process of induced-fit conformational change (see below).
Overall Structure and Folding of the GlnRS-tRNAGln-ATP Complex
The 553 amino-acid E. coli glutaminyl-tRNA synthetase is a monomeric enzyme of 63 kDa molecular weight.22-24 The enzyme possesses an elongated structure spanning about 100 Å in its longest dimension, which extends from the acceptor end to the anticodon loop of the bound tRNAGln(7) (for a detailed review see Rould & Steitz).6 Most of the structure can be readily partitioned into four discrete domains (fig. 1). The amino-terminal portion of the enzyme contains the active site, and folds into a Rossmann dinucleotide fold consisting of a six-stranded parallel β-sheet flanked by a-helices. Five of the β-strands alternate with α-helices along the primary sequence in the canonical manner,25 while the sixth is found within a connective, mainly helical subdomain (amino acids 271-339) that spans the active-site and the C-terminal anticodon-binding region. Interestingly, the polypeptide between β-strands 5 and 6 of the fold adopts an unusual left-handed connection. 9 The Rossmann fold is divided into two halves by a 110 amino-acid mixed α/β domain (amino acids 103-211) inserted between the third β-strand and third α-helix. This inserted “acceptor-binding” domain binds the 3'-end of the tRNA, which adopts a hairpinned conformation necessary for precise orientation of the terminal A76 ribose sugar into the active site. In addition to binding sites for ATP and glutamine, the Rossmann fold also contains the key Asp235 side-chain, which makes direct and water-mediated sequence-specific hydrogen bonds in the tRNA acceptor-stem minor groove at G2-C71 and G3-C70.7,16

The C-terminal portions of GlnRS serve to properly orient the tRNA on the enzyme surface, and to make sequence-specific interactions with each of the three anticodon bases: C34, U35 and G36.8 Within the helical subdomain, an a-helix-turn-β-strand-α-helix motif conserved among class 1a and class 1b synthetases interacts with the inside corner of the tRNA L-shape, apparently specifying the global juxtaposition of the two macromolecules.26 The second helix of this motif runs down the coaxial D and anticodon stems of the tRNA, bridging into two β-barrel domains that bind the anticodon loop. In primary sequence, the barrel more distal to the active site is inserted between the first two strands of the proximal barrel. A long antiparallel β-ribbon (amino acids 472 to 493) emerges from the proximal barrel, packing onto amino acids in the helical subdomain that are in direct contact with active-site peptides. Speculations regarding the mechanism by which anticodon recognition is communicated to the active site have focused on this β-ribbon, as well as on a long α-helix which binds along the tRNA from the inside corner of the L-shape to the anticodon stem/loop region (see below).7,27
GlnRS has served as a model system to investigate the folding of multidomain proteins.28,29 These studies employed urea-induced denaturation in conjunction with binding of small-molecule probes, sulfhydryl reactivity, and UV, CD and fluorescence spectroscopy. An important conclusion is that the enzyme folds to the native state through a compact molten globule intermediate resembling those previously characterized for small single-domain proteins.
Discrimination Among tRNAs
Acceptor-Stem Recognition
The earliest studies of tRNA discrimination by E. coli GlnRS predate both cloning of the glnS gene23 and determination of the crystal structure.7 In the early 1970s, genetic selections were established for the misacylation of supF tRNATyr, by requiring suppression of amber mutations at positions in which tyrosine insertion results in a nonfunctional protein product. These studies showed that mutations in this suppressor tRNA at position 73 and at acceptor-stem base-pairs 2-71 and 1-72 resulted in misacylation with glutamine by the GlnRS enzyme.30-34 Subsequently, three separate single-site mutations in GlnRS were isolated by virtue of their ability to acylate the wild-type supFtRNATyr with glutamine in vivo.16,35 These mutations alter Asp235 of the wild-type enzyme to asparagine and glycine, and Ile129 to threonine. Similar results were obtained in vivo with the D235V and D235A mutants, while much weaker suppression is observed in the case of D235K or D235E.36 Interestingly, overexpression of wild-type GlnRS in vivo will also lead to misaminoacylation of supF,37 but concomitant overproduction of tRNAGln abolishes this mischarging.38 This shows that maintenance of the proper levels of tRNAGln and GlnRS in vivo is critical to the fidelity of the aminoacylation reaction. In accordance with this notion, a tRNASer molecule was subsequently converted to tRNAGln identity in vivo by removal of tRNASer identity elements in the acceptor stem of the molecule.39 Several other studies of in vivo suppression provided further documentation that synthetase competition helps ensure the accuracy of aminoacylation.40,41
The crystal structure of the GlnRS-tRNAGln—ATP ternary complex provided a detailed view of the interactions at the macromolecular interface, and allowed correlations to be made with these genetic data. The 3'-acceptor end of the tRNA adopts a hairpinned conformation when bound to GlnRS, such that the terminal ribose of A76 is positioned in the active site adjacent to the α-phosphate of ATP (fig. 2). The base of C74 is flipped out to bind in an enzyme pocket within the inserted acceptor-binding domain, allowing colinear stacking of the G73, C75 and A76 bases. The U1-A72 base-pair is broken in the complex, and the side-chain of Leu136 stacks between the A72 ring and the G2-C71 base-pair. This unusual conformation is stabilized by extensive protein interactions as well as by an intramolecular hydrogen-bond donated by the exocyclic NH2 group of G73 to the A72 phosphate.

The structure in this region shows clearly how protein-induced conformational change and indirect readout operate to generate specificity. The requirement for disruption of the 1-72 base-pair provides discrimination against tRNAs possessing either G1-C72 or C1-G72, as the additional Watson-Crick hydrogen-bond should render these pairs more difficult to break. Similarly, only G73 can provide the 2-NH2 moiety to form the intramolecular hydrogen-bond within the RNA hairpin, allowing selectivity against tRNAs possessing any other base at this position. These findings are entirely consistent with the genetic data described above. For example, the G1-C72 base-pair of supF tRNATyr was altered to A1-C72 or to A1-U72 in the mutants which could be mischarged by GlnRS.34
The interactions observed in the crystal structure at base-pairs G2-C71 and G3-C70 are also consistent with the genetic data. At G2-C71, the exocyclic amino group of guanine donates a hydrogen-bond to the backbone carbonyl oxygen of Pro181 within the acceptor-binding domain. G2 also makes a hydrogen-bond to a key buried water molecule in the minor groove; this water also bridges to the backbone amide of Ile183, the exocyclic O2 of C71, and a carboxylate oxygen atom of Asp235. Asp235, which emanates from the amino end of an α-helix in the second half of the dinucleotide fold, also accepts a hydrogen bond from the 2-NH2 group of G3. Thus, direct and water-mediated recognition of G2-C71 and G3-C70 is tightly coupled. Since the D235 carboxylate group directly mediates specific contacts, the reduction in tRNA selectivity in vivo arising from the D235N and D235G mutants is easily rationalized, at least qualitatively.7,16 In an attempt to understand recognition at G3-C70 at a more detailed level, the crystal structures of GlnRS D235N and D235G bound to tRNA and ATP were determined. The D235N structure showed that the Asn235 side-chain makes an additional hydrogen-bond with the exocyclic O2 group of C70.20 Modelling based on the structures suggests that a similar new contact with the N3 group of A70 in the U3-A70 pair of supF tRNATyr should also be possible, providing one rationale for the relaxed discrimination of the mutant. Although these Xray studies provide important insight, structures of wild-type and D235N complexes of GlnRS bound to supF, together with a more rigorous kinetic evaluation, would clearly also be of value in understanding the biochemical basis for mischarging. Surprisingly, neither the D235N nor D235H mutants are significantly decreased in steady-state aminoacylation of the cognate tRNA.13,14
The mischarging phenotype arising from the I129T mutation is less easily explained. Since the Cγ atom of I129 is located approximately 3.6 Å from the tRNA C74 phosphate, one possibility is that the mutation to threonine might allow additional stabilization of the required hairpin structure even in the absence of specific identity nucleotides at positions 1-72 and 73. A crystal structure of I129T GlnRS bound to tRNAGln and ATP showed no significant structural changes compared with the wild-type enzyme.20
A set of biochemical and genetic experiments conducted after the structure became available further support the importance of the acceptor-end interactions. In one study altered tRNA2Gln species were synthesized by in vitro transcription and analyzed for their ability to serve as substrates for GlnRS in biochemical activity assays.42 In vitro-synthesized tRNAGln lacking modifications is less stable to thermal denaturation, but shows kinetic parameters indistinguishable from those of tRNA produced in vivo.17,43 tRNAGln mutants at G73, U1-A72, G2-C71 and G3-C70 were decreased in aminoacylation activity by factors ranging from 2 to 2000-fold, supporting the notion that the acceptor-stem interactions are important to recognition.43
Mutational experiments of the enzyme at the acceptor-end binding interface were also performed. Saturation mutagenesis was used to introduce alterations in two enzyme surface loop regions within the inserted acceptor-binding domain, at amino acids 126-138 and 178-188.44 These encompass the amino acids stabilizing the broken U1-A72 pair and hairpinned acceptor end, and those interacting at position G2-C71, respectively. GlnRS mutants with decreased tRNA selectivity were then isolated in vivo by misacylation of noncognate amber suppressor tRNAs. This study showed that the interactions made by Arg130 and Glu131 with the hairpinned 3'-acceptor end are crucial to efficient rejection of noncognate tRNAs. This is also the case for the Leu136 side-chain which stacks directly between G2 and A72. The L136F mutant constructed by site-directed mutagenesis is less specific than wild-type GlnRS both in vivo and in vitro, again showing that precise acceptor-end interactions are central to tRNA selectivity.45
Finally, a combination of chemical synthesis and transcription was used to specifically introduce inosine substitutions at nucleotides G2, G3, G5 and G10.46 The most important finding here is that the G10I mutant is decreased by 300-fold in kcat/KM for aminoacylation, a reduction comparable to tRNA mutants at G2 and G3. This suggests that the D-stem interface is also an important recognition region. A specific interaction between the N2 of G10 and Glu323 of the connecting helical subdomain in GlnRS is observed in the crystal structure, suggesting that G10 may also be important to identity in vivo. Mutation of Glu323 to Gly yielded a mutant enzyme with altered tRNA specificity in vivo as well as 100-fold reduced kcat/KM in vitro, further supporting the importance of this interaction.47
Anticodon Loop Recognition
As in the case of the acceptor stem, early genetic experiments involving in vivo mischarging of supU tRNATrp first defined the importance of the central anticodon nucleotide U35 in tRNAGln identity.48-51 The role of U35 was also shown by mutation of the tRNAfMet anticodon from CAU to CUA, which led to mischarging of this noncognate tRNA by GlnRS.52-55 In a third genetic study, overproduction of GlnRS in vivo was required for missense suppression of a CGA arginine codon, by a tRNAGln species containing C35.56 Again, the structure of the complex offers important correlations with these data. It shows that the tRNAGln anticodon loop adopts a conformation in which all three anticodon bases are unstacked to bind in complementary pockets formed by the C-terminal β-barrels of the enzyme (fig. 3).7,8 This is in sharp contrast to the cystal structures of unliganded tRNAPhe and tRNAAsp, in which all the anticodon loop bases stack on each other.57,58 The anticodon stem of tRNAGln bound to GlnRS is also extended by two non-Watson-Crick base pairs: 2'O-methyl-U32:Ψ38 and 2-methyl-A37:U33. Only one direct hydrogen-bond links each of the two pairs, but a network of ordered water molecules assist in stabilizing the base-pairing configuration. The transposed N5 nitrogen of pseudouracil is involved in these water networks, as shown also by comparisons of structures determined with in vivo vs. in vitro-derived tRNA.17 The two new base-pairs exhibit A-form geometry and stack well with the anticodon stem. These expected deformations of the tRNAGln anticodon loop provide a second example of an important induced-fit conformational change in the tRNA, in which a presumably higher-energy conformation of the RNA is stabilized by means of extensive protein contacts.
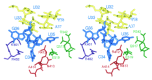
Each of the three anticodon bases is recognized primarily by a separate single stretch of polypeptide, although other protein residues also form part of each pocket.8 For each of the three bases C34, U35 and G36, a basic residue from the enzyme makes a salt bridge with an adjacent phosphate group, and the aliphatic portion of the Arg or Lys packs against either the sugar or hydrophobic surface of the base. Base recognition involves both main-chain and side-chain protein hydrogen bonds with the Watson-Crick moieties (fig. 3). Specific side-chain interactions with U35 are made by Arg341 and Gln517, correlating with the extensive genetic data, while specific side-chain/base contacts with G36 are made by Arg402. Interestingly, although GlnRS must recognize both the CUG and mnm5s2UUG anticodons, the crystal structure also shows highly specific contacts with C34. Both the N3 and exocyclic 4-NH2 groups of C34 make specific hydrogen bonds to the main-chain at Asn413 and Ala414 of GlnRS. This finding implies that binding of the tRNA1Gln mnm5s2UUG anticodon to GlnRS requires a conformation of the protein that is distinct from that observed in the known crystal structures. A segment of the GlnRS enzyme adjacent to the C34 binding site (amino acids 443-453) is disordered in all the crystal structures determined,8 and it is possible that this peptide is involved in the recognition of tRNA1Gln as well as the discrimination against noncognate species, some of which also possess bulky modified groups at the wobble position. The C-terminal hexapeptide of GlnRS (amino acids 548-553) is disordered in the structures as well, and is similarly located directly at the interface with the anticodon loop.
As demonstrated for the acceptor-end interactions, the importance of anticodon nucleotides was also shown by in vitro aminoacylation assays of tRNA mutants obtained by in vitro transcription.42 Large decreases in kcat/KM ranging from 60 to 30,000-fold were observed for substrates mutated separately at positions C34, U35, G36, A37 or U38. Further, the mutant GlnRS enzymes R341A and R402A are significantly decreased in catalytic rate, which also supports the importance of the U35 and G36 interactions (fig. 3).47 The recognition of tRNA1Gln, which possesses the modified nucleoside 5-methylaminomethyl-2-selenouridine at the wobble position,59 has also been examined. Treatment of tRNA1Gln with cyanogen bromide resulted in a 10-fold reduction in apparent binding affinity.60 More recently, a comparison was made of the aminoacylation efficiency of the unmodified tRNA1Gln transcript, with that of tRNA1Gln derived in vivo.61,61a This showed that the transcript is increased 10-fold in KM and decreased 4-fold in kcat compared with the modified species. Since the RNA2Gln transcript is not compromised in aminoacylation, and the U34 derivative is the only modified base unique to tRNA1Gln> (fig. 1B), it may be concluded that the modified portion of mnm5s2U34 is contributing significantly to recognition by GlnRS. The presence of a U31-A39 base-pair in tRNA1Gln, rather than the A31-Ψ39 of tRNA2Gln, might also be significant to recognition of the tRNA1Gln anticodon loop.
Indirect Readout
The determination of the GlnRS crystal structure,7,8 and the tRNA mutational experiments immediately following,42,46 led to the proposal that a “complete identity set” had been elucidated for the E. coli glutamine system. The set consisted of the following 15 nucleotides: G73, U1-A72, G2-C71, G3-C70, G10, and all seven nucleotides of the anticodon loop.46 The set was hypothesized based on a combination of evidence from the crystal structure (all 15 nucleotides), in vivo genetics (nucleotides G73, U1-A72, G2-C71 and U35), and in vitro biochemical studies (all 15 nucleotides). All of the interactions made by GlnRS with discriminating base functional groups are included with the exception of a peripheral contact at C16, which appears unimportant based on in vitro mutagenesis.46 With reference to the structure it is clear that the proposed identity elements might act either directly with amino acids of GlnRS (G2-C71, G3-C70, G10, C34/U34, U35, G36), or indirectly to facilitate an RNA conformation required for aminoacylation (G73, U1-A72, Um32, U33, m2A37, Ψ38).
Testing whether these nucleotides are indeed sufficient to specify tRNAGln identity requires their transplantation into noncognate tertiary frameworks. Two such experiments have been carried out. Glutaminylation of a tRNAAsp mutant possessing all 15 proposed tRNAGln identity determinants showed that this tRNA is indeed an efficient GlnRS substrate, with comparable kinetic properties to wild-type tRNAGln.62 Interestingly, however, when tRNAGln-specific nucleotides in the D and variable loops were further added to the fully functional hybrid Asp/Gln tRNA, glutaminylation then decreased by 25-fold. In the other experiment, introduction of 14 tRNAGln identity nucleotides into E. coli tRNAGlu produced a hybrid tRNA which was decreased 100-fold in GlnRS substrate efficiency compared to wild-type tRNAGln.61 This tRNA possessed C32 as the sole non-identity nucleotide among the hypothesized set of 15.46 The omission might be important, because the unprotonated N3 of C32 would disrupt the native U32-Ψ38 hydrogen-bond, thus destabilizing the required anticodon loop conformation [mutation of Ψ38 to G38 in tRNAGln causes an identical 100-fold decrease of activity]. 42 When the entire tertiary core of tRNAGln was also introduced, aminoacylation remained 25-fold below that of the cognate substrate. Thus, either the C32-U38 mispair, or tRNAGlu base-pairs in the anticodon and acceptor stems, must be responsible for the decreased GlnRS activity.61
While confirming the importance of the acceptor-end and anticodon nucleotides, these experiments also suggest that other parts of the tRNA structure have significant effects on GlnRS recognition. Thus, nucleotides in the tertiary core, the anticodon stem, and the bottom portion of the acceptor stem may also need to be considered as part of the glutamine tRNA “identity set”. Indeed, a number of other experiments have directly shown the importance of these portions of the substrate. In vivo suppression studies have demonstrated important roles for the base-paired D-stem,11,63,64 the G15-C48 and A13-A22 base interactions in the tertiary core together with the bottom portion of the acceptor stem,11,64 and the anticodon stem.65
The importance of several D-stem base-pairs was first shown by their requirement for suppression by an ochre T4 phage tRNAGln species.63 By contrast, a U30-G40 base-pair in a yeast amber tRNAIle molecule allowed GlnRS suppression while introduction of the G30-C40 tRNAGln pair into this noncognate species abolished the effect.65 However, most of the in vivo evidence for an expanded identity set derives from experiments with a mutated tRNAAla amber suppressor which does not insert alanine in vivo and which inserts glutamine only weakly.11,64 Further mutations in the tRNA were then sought which make this starting species a better GlnRS substrate. By this strategy the following nucleotides in the interior of the molecule were identified as determinants of GlnRS recognition in vivo: G4-C69, G5-C68, C11-G24, A13-A22, G15-C48 and U32-Ψ38. The mutants create mispairs including the G-U wobble pair at these positions, presumably influencing the local tRNA structure in their vicinity. Strikingly, GlnRS interacts with the sugar-phosphate backbone, but not the bases, at each of these nucleotides. This demonstrates clearly that specific GlnRS recognition requires interactions with tRNA backbone groups whose precise positions are ultimately determined by the base sequence.
The importance of indirect readout of tRNA sequence information by GlnRS has also been shown by in vitro studies. A set of tRNAGln variants possessing the large variable loops of class II tRNAs were constructed and analyzed for thermal stability and glutaminylation kinetics.12,43 At least four distinct class II core region folds, all containing the 20-nucleotide variable arm of T. thermophilus tRNASer, were identified based on phylogenetic analysis and incorporated into the tRNAGln molecule. These studies showed that GlnRS can efficiently glutaminate tertiary core variants of every structural class, with kcat/KM values up to 25% of wild-type tRNAGln. However, the kinetic analyses also revealed a significant dependence on the number and identity of the nucleotides inserted into key portions of the core. Strikingly, GlnRS requires one or two unpaired uridines 3' to the variable arm to efficiently aminoacylate several of the class II frameworks. Overall, kcat/KM values vary across a 200-fold range for substrates with plateau aminoacylation values sufficiently high for reliable analysis. It is thus clear that the contribution of the core region is quite comparable to that of many previously identified recognition elements in the acceptor stem and anticodon.42,46
Thus, while some studies have shown limited effects of core region mutation on GlnRS function,13,18 it is now clear that an important aspect of discrimination arises from the rejection of noncognate tRNAs possessing noncomplementary cores. Because GlnRS interacts only with the inside of the tRNA L-shape, a likely mechanism involves propagation through the tRNA structure.12 The variable arm interacts with the D and augmented D-stems on the major groove side, thus helping to determine the detailed architecture of the helix and the consequent positioning of sugar-phosphate backbone moieties. By binding on the opposite minor groove side of the stem, it appears likely that GlnRS then discriminates against an altered core by making suboptimal contacts along this and adjacent portions of the interface. Subtle modulation of these interactions likely also arises when base-pairs identified in the in vivo studies are altered.11,64 The mechanism of indirect readout is closely tied to that of the induced fit conformational changes which occur in both tRNA and GlnRS upon complex formation, and depends on a variety of energetic factors which come into play during the course of forming the catalytically proficient active site (see below).
The core region of tRNAGln may be optimized to promote efficient catalytic turnover. A library of tRNAGln molecules randomized across the variable core domain was constructed and subjected to in vitro selection to improve binding to GlnRS and to EfTu.66 Based on the sequence analysis of tight-binding aptamers from this library, a four-nucleotide 5'-AGGU variable loop was inserted into tRNAGln, replacing the native five-nucleotide 5'-CAUUC loop. Gel retardation analysis demonstrated that the mutant tRNA binds 50-fold more tightly than the wild-type species, but the crystal structure of the aptamer bound to GlnRS showed that there were no differences in protein-RNA contacts across the entire interface.19 Thus, it appeared that the tighter binding affinity arises from a decreased entropic cost of binding, which is associated with a better-packed core region of the mutant. The aptamer core features a set of new tertiary interactions compared with native tRNAGln, including an unusual trans wobble G-U base-pair. Presumably, optimal efficiency on the enzyme has selected for a native tRNA which binds more weakly, possibly to ensure rapid product release. The ability of GlnRS to adapt to altered core regions is also shown by crystal structures of the enzyme bound to mutants containing the unusual G15-G48 Levitt pair of tRNACys.18 This study also showed the capacity of the Levitt pair to adopt different hydrogen-bonding interactions depending on the surrounding tRNA structure.
Amino Acid Specificity
Beyond the observation that GlnRS discriminates strongly in favor of glutamine, little information regarding the mechanism of amino acid specificity was available until the first X-ray structures were determined. None of the initial structures were solved bound to the glutamine substrate. However, the structural similarity of the GlnRS Rossmann fold to that of TyrRS bound to tyrosyl adenylate,67 as well as the observation of an empty pocket in GlnRS situated in an analogous position adjacent to the ATP, allowed prediction of the general binding orientation.9 This was later confirmed by determination of the GlnRS crystal structure at 2.4 Å resolution bound in a ternary complex with tRNAGln and 5'-O-[N-(L-glutaminyl)sulfamoyl]adenosine (QSI).10 QSI is an analog of glutaminyl adenylate in which the O-P-O phosphodiester is replaced by N-S-O, such that a secondary amide—NH group replaces the oxygen which bridges to the carboxyl carbon of the amino acid. This inhibits the second step of aminoacylation, because (unlike the bridging phosphate oxygen of AMP) the bridging nitrogen in the sulfamoyl link will not readily depart as the leaving group. The Ki of QSI for the GlnRS reaction was determined to be 1.3 μM.10 This is about five-fold weaker than that of the related analog glutaminol adenylate, which possesses a Ki of 0.28 μM.68
QSI binds in the active site cleft adjacent to the 3'-terminal ribose of tRNA and makes interactions with amino acids from both halves of the Rossmann fold (fig. 1). Structural comparisons showed that the common adenosine and ribose moieties of ATP and QSI bind nearly identically to GlnRS. Further, the interactions of the QSI sulfamoyl group and the ATP α-phosphate are also very similar. The 2'-OH of tRNAGln A76 plays the role of hydrogen-bond acceptor from the bridging —NH of QSI, similar to its interaction with the a-phosphate of ATP.9,10 The α-NH3+ group of QSI donates three hydrogen bonds: to the 3'-OH of tRNAGln A76, to the mainchain carbonyl oxygen of Pro32, and to the side-chain carboxylate group of Asp66. Thus, both 3'-terminal hydroxyl groups of tRNA help to orient glutaminyl adenylate in the active site.
A surprising feature of the QSI cocrystal structure is the absence of direct, strongly discriminating interactions with the amide side-chain of glutamine (fig. 4A). The amide oxygen makes no hydrogen-bonding interactions with the enzyme. Further, the two hydrogen-bonds donated by the amide —NH2 group (which distinguishes glutamine from glutamate) are to groups with ambiguous hydrogen-bonding character. Therefore, in order to generate specificity, it appeared that both the water molecule and the Tyr211 side-chain making direct contacts must be oriented by other moieties. Remarkably, the network of interactions apparently necessary to ensure this orientation extends to include four waters and the participation of seven main-chain and side-chain groups, all emanating from the second half of the Rossmann fold (fig. 4A). These water-mediated hydrogen-bonding interactions observed in the QSI complex are also present when free glutamine is bound.120 The latter structure was obtained by cocrystallization of GlnRS with tRNAGln, glutamine and the ATP analog AMPCPP.21
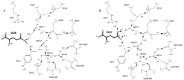
Based on these observations, it appeared that the exclusion of glutamate must necessarily be based on an unfavorable interaction of the carboxylate side-chain with the oriented hydrogen-bond acceptors of Tyr211 and the adjacent water. However, a cocrystal structure of the GlnRS-tRNAGln complex bound to noncognate glutamate shows that this is not the case.120 In that structure cocrystallized with AMPCPP, glutamate binds in an alternate position in which the side-chain carboxylate makes an ion-pair interaction with Arg30 (fig. 4B). Further, compared to the analogous quaternary complex bound to glutamine, the α-carboxylate group of glutamate is mispositioned with respect to the tRNAGln terminal ribose. These observations suggest that the GlnRS amino acid pocket discriminates by accommodating the structurally similar noncognate substrate in a nonproductive binding orientation. Interestingly, the glutamate side-chain carboxylate group also accepts a hydrogen-bond from Tyr211, reversing the directionality of this interaction compared with the glutamine substrate. Because there are no other structural changes in the binding pocket, this shows that the water-mediated network of hydrogen-bonds is capable of accommodating either a donor or acceptor moiety to Tyr211. Presumably, this occurs by small reorientations of the intervening water molecules (fig. 4B). It appears then that the orientation of the glutamine amide group may not be fixed in the substrate binding pocket, and that specificity arises entirely from the steric exclusion or nonproductive orientation of noncognate side-chains in the pocket. This finding is of interest to consider in light of the possible evolution of GlnRS from a nondiscriminating glutamyl-tRNA synthetase capable of aminoacylating both tRNAGln and tRNAGlu with glutamate.69-71
The QSI cocrystal structure also showed that the 3'-terminus of the tRNA participates in formation of the glutamine binding site. It does so via the direct interactions with A76 ribose hydroxyls as mentioned above, and also by stacking of the A76 adenine ring onto Tyr211 and Phe233.10 Mutational analysis supports the involvement of Asp66, Tyr211 and Phe233 in glutamine binding.72 Conservative replacements in these side-chains resulted in 20 to 60-fold elevations in the glutamine KM, without significantly compromising the kcat for aminoacylation.
Two studies have been published in which attempts to alter the substrate specificity of GlnRS toward glutamate were made.73,74 In the first of these, a genetic screen for amino acid mischarging was established based on suppression of a mutation in β-galactosidase.73 Mutants at Tyr240 and Phe90 of E. coli GlnRS were isolated and characterized for mischarging in vivo and in vitro. Little incorporation of Glu into the third position of DHFR expressed in vivo was detected. The purified mutants did show 3-5 fold reduced Ki values for glutamate in the glutaminylation reaction, but as the glutamine KM values were also reduced it appeared that little specificity alteration was achieved. Neither side-chain is located within the amino acid binding pocket.
The second study was carried out on the human GlnRS enzyme.74 Site-directed mutagenesis was used to introduce mutations at positions equivalent to Cys229 and Gln255 of E. coli GlnRS, each of which is located within the amino acid binding pocket. It was found that the double mutant C229R/Q255I was improved in glutamyl-adenylate formation by six-fold and in glutamylation of tRNAGln by 45-fold, compared with wild-type GlnRS. However, the mutants remain compromised by approximately 104-fold in catalytic efficiency compared to the specific reactions of wild-type GlnRS. The large changes in relative kcat/KM values for the mutants are mainly accounted for by decreases in their glutaminylation activities rather than improvements in glutamylation. The inability to significantly alter GlnRS amino acid specificity by a small number of local mutations suggests that amino acid determinants responsible for evolution of the glutamine binding site are more broadly dispersed in the enzyme structure.
A different approach toward engineering the GlnRS amino acid pocket relies on genetic selection of a misacylating GlnRS, which would incorporate a nonstandard amino acid into proteins in vivo through use of an “orthogonal” tRNA-synthetase pair. Two such pairs which incorporate GlnRS have been reported. One of these utilizes S. cerevisiae GlnRS together with S. cerevisiae tRNAGln, for use in E. coli.75 The second pair features E. coli GlnRS together with an amber suppressor derived from human initiator tRNA, for use in yeast.76 Either of these pairs might form the basis for genetic selections designed to isolate GlnRS mutants which incorporate nonstandard amino acids. Attempts were also made to engineer the E. coli GlnRS-tRNAGln pair for use in E. coli, but selections employed in this case, for isolation of an orthogonal tRNAGln, did not succeed in isolating mutant enzymes which discriminate against wild-type E. coli tRNA2Gln.75,77 The two orthogonal pairs rely on nonsense suppression, which produces truncated proteins as byproducts in the cell. Conceivably, a missense suppression system involving GlnRS suppression of CGA (a rare arginine codon) might alleviate this potential difficulty.56
Enzymatic Mechanism
Refinement of the respective GlnRS cocrystal structures with ATP and glutamine bound have provided considerable insight into the stereochemical mechanisms of aminoacylation.9,10 The structure is in a conformation similar to that of the active enzyme in solution, as shown by the ability of the crystalline ternary complex to catalyze aminoacyl adenylate formation when glutamine is soaked into the crystals. Further, the 2'-OH of the terminal A76 ribose of tRNAGln is positioned in the active site, where it donates a hydrogen bond to the α-phosphate of ATP. This is consistent with biochemical studies indicating that aminoacyl transfer is to the 2'-OH position, as found for all class I tRNA synthetases.3
Amino Acid Activation
ATP makes a number of interactions with residues in a β-strand-turn-α-helix motif at the amino terminus of the first half of the Rossmann fold, and some contact as well with amino acids in the second half (fig. 5).7,9 To a first approximation ATP and glutamine occupy positions on opposite sides of the Rossmann fold pseudo-dyad axis. The specificity for ATP is ensured by Watson-Crick-like hydrogen bonds of the adenine ring with the backbone amide of Leu261. The adenine ring rests on the polypeptide backbone at Gly42 of the conserved class I HIGH motif, providing a rationale for the conservation as Gly, since a β-carbon would block this contact. The Arg260 side-chain stacks on the opposite side of the adenine ring, and its' guanidinium group makes a water-mediated contact to the ATP α-phosphate. The imidazole rings of HIGH interact with the local polypeptide backbone such that the protonated nitrogens are each directed toward the ATP phosphates. However, the His40 imidazole also donates a hydrogen-bond to the backbone carbonyl group of Met268 from the conserved MSK motif, while the Met side-chain packs under the His40 imidazole. These mutual interactions provide a convincing rationale for the simultaneous presence of HIGH and MSK among all the class I synthetases. Further, Lys270 of MSK donates two hydrogen bonds to the ATP α-phosphate and is stabilized by a third with the side-chain of Asn36. Other hydrogen-bonds to the ATP are made by Thr230, Ser 46 and Leu228 (to the ribose sugar), and by Glu34 and Asn36 (to the phosphates). Amino acids N and C-terminal to MSK make hydrophobic packing interactions with residues which form part of a large loop connecting two antiparallel β-strands of the proximal β-barrel domain, providing a structural connection from the active site to the anticodon-binding region of the enzyme.
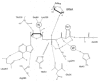
In contrast to the wealth of structural detail available, there have been few enzymological studies of the GlnRS catalytic mechanism. It is known that the rate of glutaminyl adenylate formation is significantly faster than that of aminoacyl transfer to the tRNA. This indicates that Gln-tRNAGln cannot be an obligatory intermediate in the synthesis of glutaminyl adenylate, suggesting that the enzymes' requirement for tRNA to catalyze ATP/PPi exchange arises from RNA-induced conformational changes in the active site.78 It is known that yeast tRNA and human tRNA are equally capable of promoting the ATP/PPi exchange reaction of human GlnRS, despite the fact that heterologous yeast tRNA is only weakly active in aminoacylation.74 This suggests that global RNA-protein interactions play an important role in inducing active-site formation. Further, a study of E. coli GlnRS indicated that tRNAGln containing a 2'-deoxy modification was unable to activate the enzyme for ATP/PPi exchange,3 implying that the 2'-hydroxyl group may directly facilitate this reaction. The recent crystal structure of unliganded E. coli GlnRS reveals that a number of amino acids binding both glutamine and the hairpinned tRNA acceptor end are significantly mispositioned with respect to the remainder of the active site.121 This shows directly that tRNA binding is coupled to construction of a catalytically proficient active site.
Despite the absence of rigorous enzymological studies, detailed proposals for the stereochemical mechanism of glutaminyl adenylate formation have nonetheless been deduced from the crystal structures, and are consistent with the wealth of biochemical data available for the reaction of the homologous class I TyrRS. The structure is consistent with the known in-line displacement mechanism of amino acid activation by TyrRS, since the glutamine carboxylate is positioned on the opposing side of the ATP'-phosphate relative to the pyrophosphate leaving group.9,10 Also, there do not appear to be appropriately positioned residues which might be directly involved as acid-base or covalent catalysts, which is consistent with TyrRS kinetic studies indicating that the role of the enzyme is solely to bind and orient the ATP and amino acid substrates.79 To assess the roles of individual GlnRS amino acids in detail, the pentacovalent transition state at the ATP a-phosphate was modeled based on the ATP cocrystal structure. This modelling study suggested that His43 and Lys270 from the conserved class I motifs are each involved in preferential transition state stabilization, as their interactions with the α-phosphate improve as the geometry of this reactive group changes from ground-state tetrahedral to transition-state trigonal bipyramidal.9 This is consistent with studies of mutants at the equivalent His48 and Lys233 amino acids of B. stearothermophilus TyrRS, which showed that these side-chains preferentially stabilize the transition state in that enzyme.79,80
To locate divalent metal ions in the GlnRS active site, crystals of the enzyme were soaked in solutions containing manganese ions and a data set to 4 Å resolution was obtained.9 A manganese ion was located bridging the α and β-phosphates of ATP on the face opposite to nucleotide A76 of the tRNA. Since GlnRS can catalyze reactions using Ca2+, Mn2+, Co2+ or Mg2+ cations,81 and because there are not stringent geometric constraints at this position, this may represent the site bound by any divalent metal ion which supports GlnRS catalysis. The metal at this site can assist the reaction by withdrawing electrons toward the β-phosphate and possibly by facilitating a productive conformation of the pyrophosphate. This metal site is similar to that described for some other tRNA synthetases, including TyrRS.82
Synthesis of Gln-tRNAGln
Two proposals exist for the mechanism of the second step in aminoacylation-the transfer of glutamine from the adenylate intermediate to the 2'-OH group of tRNAGln A76. It was first suggested that the phosphate of glutaminyl adenylate serves as the base to abstract the proton from the 2'-OH of A76, as the oxygen nucleophile attacks the carbon of the mixed anhydride intermediate.9 However, this proposal was criticized because the pKa for this phosphate is approximately 1.5-2.0, similar to that of a phosphodiester in DNA.10 An alternative proposal is that the nearby carboxylate of Glu34 functions as a base, but indirectly through a water molecule.10 This appears difficult chemically, because the high pKa of water disfavors ionization at physiological pH in the absence of an adjacent divalent metal ion or other strongly polarizing group. From a biological perspective, the idea is also problematic because Glu34 is not conserved among class I synthetases. The original proposal gains merit when it is recognized that the phosphate of the AMP product protonates at 6.5-7.0. Thus, as the second step of the reaction begins and the charge distribution changes in the moieties poised for catalysis, the pKa of the adenylate phosphate begins to rise. This renders it more likely to accept the proton than previously thought.9 A direct role for the phosphate is also consistent with greatly decreased rates of tRNA aminoacylation by PheRS and MetRS, when phosphorothioate ATP analogs are substituted for ATP.83,84 The idea of intramolecular catalysis for the second step of aminoacylation is attractive because it can be generalized to all tRNA synthetases, and because it suggests the feasibility of a primordial RNA-based tRNA synthetase which lacked protein groups. However, additional experiments to evaluate these two mechanisms are still needed to resolve the issue.
A different proposal with respect to the mechanism of the second reaction is that Arg260, which makes a water-mediated interaction with the β-phosphate of ATP, may play a role in stabilizing the carbonyl oxygen of glutaminyl adenylate during nucleophilic attack on the carbon.10 However, this proposal is not supported by the results of site-specific mutagenesis: the kcat of the R260S mutant for tRNA aminoacylation is within two-fold of wild-type GlnRS.14 Arg260 may instead be more directly involved in ATP and glutamine binding, since the KM values for these substrates are elevated in the mutant enzyme.
There is also evidence that E. coli GlnRS is phosphorylated in vivo in a process involving the heat-shock proteins DnaK and DnaJ.85,86 Three distinct forms of GlnRS possessing phosphorylated threonine residues were demonstrated by two-dimensional gel electrophoresis. This is a potentially important finding, but there have been no further studies to investigate which threonine(s) are modified or to examine whether phosphorylation modulates activity. No electron density corresponding to phosphorylated threonine is evident in the crystal structures. Interestingly, the GlnRS enzyme from rabbit reticulocytes, a component of the multi-synthetase complex in that organism, is also phosphorylated in vivo.87
Methods for Enzymological Studies
The extensive studies of E. coli GlnRS have included development of a range of methods for expression and characterization of wild-type and mutant enzymes. Superb high-level overexpression strains are available for producing both GlnRS and tRNA2Gln in E. coli.15 Additionally, a variety of strain backgrounds have been developed which are useful for working with mutants. These include strains expressing a temperature-sensitive glnS gene,3,47 as well as a strain carrying a glnS chromosomal deletion.27 A chromosomal mutant in E. coli was used to facilitate separation of wild-type and expressed mutant GlnRS enzymes, by fusing a reporter epitope to the 5'-end of the wild-type glnS gene.88 The epitope-tagged wild-type enzyme could then be removed by chromatographic separation. Low-level expression of E. coli GlnRS in S. cerevisiae has also been achieved as a fusion protein carrying an epitope tag.76,89
tRNA binding studies to GlnRS have been performed by three different methods: fluorescence spectroscopy, filter-binding, and gel retardation analysis. The fluorescence studies were performed by linking a reporter fluorophore to the enzyme through a sulfhydryl linkage. This allowed measurements of the tRNA dissociation constant (Kd), as well as the Kd values for ATP and glutamine.90,91 The measured constants by this method are as follows: Kd(tRNA) = 0.22 μM; Kd(glutamine) = 460 μM; Kd(ATP)= 180 μM. Kd(tRNA) calculated based on measuring on-rates (kon) as a function of tRNA concentration, also by fluorescence, was found to be similar: 0.33 μM.14 By contrast, the Kd value measured by gel retardation analysis using 3'-end labeled tRNA is three-fold tighter, 0.07 +/- 0.01 μM;19 this value matches that obtained by filter binding to within experimental error.66 Steady-state kcat values under standard conditions22,92 are in the range of 150-200 min-1,13,43,47,72 while KM values are similar to the Kd determinations for each substrate.
A continuous spectrophotometric method for determining GlnRS activity was recently developed which may have application for other tRNA synthetases as well.93 In this approach, the pyrophosphate generated in amino acid activation is hydrolyzed to phosphate by pyrophosphatase, which is then used as a substrate for purine nucleoside phosphorylase. This enzyme cleaves a ribonucleoside substrate to generate 2-amino 6-mercapto 7-methylpurine, which absorbs strongly at 360 nm. The initial velocity measured using this method is very similar to that determined by the conventional filter-binding method, although steady-state parameters were not determined. It was, however, demonstrated by this approach that a 1:1 stoichiometry exists between the production of pyrophosphate and tRNA aminoacylation.93
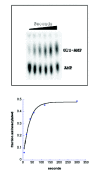
The conventional filter-binding assay for tRNA aminoacylation by GlnRS, which utilizes 14C or 3H glutamine, possesses serious limitations. The most pronounced of these was highlighted by the work of Ibba et al13 who showed that the measured steady-state kcat depends on the concentration of amino acid used in the assay. This revealed that the amino acid had typically been present in subsaturating quantities (see ref. 42 for an example), because achieving saturation is difficult given commercially available specific activities for the labeled glutamine. To address this as well as other limitations, a new assay for tRNA aminoacylation kinetics was developed (Fig. 6).94,120,122 In this approach, which should be general to most or all tRNA synthetases, full-length tRNA is first 3'-labeled with 32P using the nucleotide exchange activity of tRNA nucleotidyltransferase. Following aminoacylation, the reaction is quenched by decreasing the pH to 5.0, and the mixture is digested with P1 nuclease, which cleaves 5' to each tRNA phosphate yielding a mixture of 32P-AMP (substrate) and 32P-aminoacyl-AMP (product). These are then separated by thin-layer chromatography and quantitated by phosphorimager analysis.122 The assay is applicable to steady-state, pre-steady state and single-turnover reactions; in the latter case, it reports on the rate of the chemical step (or a closely-linked physical rearrangement after binding of all substrates). Further, by titrating concentrations of amino acid, ATP or tRNA in single-turnover reactions, Kd values may be determined kinetically. Advantages of the assay include direct measurement of the fraction of functional tRNA substrate, the ability to easily saturate amino acid concentration, and very high sensitivity. The assay has been applied to aminoacylation by both AlaRS94 and GlnRS. In the latter case, measurements of tRNAGln misacylation rates with glutamate have been possible (Fig. 6) in addition to characterization of the cognate reaction.
Induced Fit
As is the case for the formation of many macromolecular complexes, the process of tRNA recognition by GlnRS is expected to involve a set of mutually induced conformational changes. Thus, the conformation of both the enzyme and tRNA in the catalytically competent state may be envisioned as the final step in a pathway which begins with formation of an initial “encounter complex”. The free energy costs of the rearrangements, involving both entropic and enthalpic components, must be compensated for by the favorable interactions made in the final complex. Moreover, the process is also modulated by the binding of ATP and glutamine. GlnRS is one of four synthetases (along with GluRS, ArgRS and class I LysRS)71 which require the presence of tRNA to catalyze adenylate formation. This in itself may be taken as indirect evidence for an induced-fit process, since clearly the active site is not in a competent conformation until tRNA is present.
Although there is no direct biochemical evidence for conformational changes in tRNAGln upon binding GlnRS, these may be inferred with some confidence from comparisons of the cocrystal structure with the unliganded structures of other tRNAs, together with the likelihood that all tRNAs will possess similar propensities to stack acceptor-end and anticodon bases in the absence of enzyme. At a minimum, it is thus expected that tRNAGln will undergo local rearrangements at the anticodon and the acceptor end, although more subtle movements in the remainder of the molecule are also possible.
There is considerable direct and indirect evidence for protein conformational change upon substrate binding. Neutron solution scattering studies have shown significant intensity differences in the high-angle region between the unbound and tRNA-bound enzymes.95 However, these conformational changes do not involve very large-scale reorientations, since both the maximum intraparticle distance as well as the radius of gyration do not significantly change. Several fluorescence studies have also revealed conformational changes in GlnRS upon substrate binding.90,91,96 First, pyrene and bis-ANS probes were employed to show that no large-scale global enzyme reorientation occurs upon tRNA binding. 90 This is consistent with the neutron scattering work. Second, these probes were used to show that ATP binding, in the absence of other ligands, promotes a GlnRS conformational change.91 Further, the presence of ATP both quenches a small fluorescence enhancement observed upon tRNA binding alone, and alters the ionic strength dependence of tRNA binding. Together, these data suggest that ATP and tRNA binding may cause distinct rearrangements in the enzyme. Lastly, noncognate tRNAGlu does not cause the ATP-dependent conformational change observed when tRNAGln binds to the enzyme.96 This important observation suggests that the induced fit process is directly involved in tRNA discrimination. An assay which measured the extent to which tRNA was protected from metal-dependent cleavage upon GlnRS binding also suggested differences in the conformations of cognate and noncognate complexes.97
Less direct evidence for induced fit arises from activity measurements on modified tRNAs. Both tRNAGln anticodon mutants as well as core-region mutants show significant effects of up to 104-fold on kcat.12,13,42 Although kcat cannot be assigned to a particular microscopic rate constant, it is highly likely that such large decreases have a considerable component associated with the rearrangements of the complex and/or the subsequent catalytic steps (although an alternative could be that some mutants affect physical steps following the reaction). Thus, these tRNA mutations located far from the active site may be interfering with the required conformational changes, resulting in improper active site assembly and mispositioning of the reactive substrate groups.
Another set of experiments showed that the specific enzyme-tRNA contacts important to discrimination are coupled to the KM for glutamine in the aminoacylation reaction.13,14, 98 Kinetic analysis of tRNAs mutated at previously identified identity nucleotides showed that interactions at the acceptor end, anticodon loop, and D-stem each contribute significantly. An 80-fold range was measured in the glutamine KM, with most tRNA mutants causing significant increases. Given the free cellular glutamine level of 150 μM, increasing the KM above the wild-type 200 μM level by even a few-fold would cause substantial decreases in in vivo aminoacylation. Thus, misaminoacylation in vivo should be decreased by the inability of noncognate complexes to compete for glutamine binding. This represents an important additional mechanism for ensuring translational fidelity.13 The ability of tRNA contacts across the complex to affect glutamine interactions also can be taken as evidence for induced-fit conformational changes.
The existence of induced fit rearrangements raises the question of how the distal binding signals are transmitted through the structure of the complex, to affect events in the active site. Several proposals have been made along these lines. Based on the initial crystal structure of the complex, it was suggested that the signalling pathway between the anticodon and the active site involves a long antiparallel β-ribbon, which emanates from the proximal β-barrel domain to pack upon sequences adjacent to the loop containing the MSK active-site motif (Fig. 7).7 This suggestion was supported by a genetic study, in which mutants in the ribbon motif capable of improved aminoacylation activity towards an opal suppressor derived from tRNAGln were isolated.99 An alternative pathway, involving propagation of the anticodon-binding signal through the motif which binds the extreme inner corner of the tRNA L-shape, has also been proposed based on genetic data.27 The latter pathway also may operate in the propagation of signals from the core region of the tRNA.12 Finally, based on the observation that kinetic defects in tRNA mutants are generally significantly larger than those in enzyme mutants which disrupt the same interaction, it was suggested that communication occurs mainly through the tRNA structure.47 Additional evidence in support of this idea may be taken from aminoacylation studies of a tRNAGln microhelix consisting of the seven base-pair acceptor stem. The microhelix is decreased by over 107-fold in kcat/KM compared to wild-type tRNA, and its activity could not be reconstituted by addition of the anticodon stem-loop in trans.99a Thus, covalent constraints in the tRNA are important to signalling anticodon loop binding to the GlnRS active site.
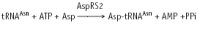
A recent computational study, which investigated both vibrational dynamics of free tRNAs and global motions within the GlnRS-tRNA complex, identified regions within the enzyme which may be involved in controlling cooperative motion upon tRNA binding.100 The calculations give information on the amplitudes of motion of domains, subdomains, and structural motifs, indicating which parts of the molecule assume enhanced mobility upon binding, and which are highly constrained. Based on this analysis it was suggested that the acceptor-binding domain and the distal β-barrel are primarily involved in tRNA recognition, having the capacity for large-amplitude global motion. By contrast, a set of amino acids clustered relatively near the active site (residues 40-45, 260-270, 306-314, 320-327, and 478-485) exhibited almost no motion and were proposed to function as a hinge-bending region controlling the cooperative transitions.100
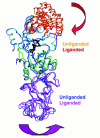
The recently determined 2.65 Å crystal structure of unliganded GlnRS now provides direct insight into the enzyme conformational changes which occur upon tRNA binding (Fig. 8).121 Acceptor-end binding of the tRNA triggers a rotation of the inserted acceptor-binding domain with respect to the core elements of the Rossmann fold, forming the binding pocket for the splayed-out C74 base as well as for the glutamine substrate. In the unliganded enzyme, the binding site for glutamine is not fully formed, explaining why tRNA is required for ATP-PPi exchange. A complex set of conformational changes also link the anticodon-binding β-barrels with the active site. It is thus clear that both motifs play an important role in the intramolecular communication.
Evolution of Glutaminyl-tRNA Synthetase
Interest in the structure-function relationships of E. coli GlnRS is heightened by the different mechanisms used to synthesize Gln-tRNAGln in different organisms across the three domains of life. In the eukaryotic cytoplasm and in some bacteria (eg, E. coli), Gln-tRNA is directly synthesized by GlnRS.71,101 However, all archaebacteria, a majority of eubacteria, and most eukaryotic organelles (the Leishmania tarentolae mitochondrial GlnRS is an exception)102 instead synthesize Gln-tRNAGln by a two-step pathway. First, a nondiscriminating GluRS misaminoacylates tRNAGln with glutamate. Second, a tRNA-dependent Glu-tRNAGln amidotransferase (Glu-AdT) amidates the noncognate complex to form the correct Gln-tRNAGln product.70 In terms of evolutionary history, it is believed that eukaryotic GlnRS is derived from a nondiscriminating GluRS, and that the presence of GlnRS in some prokaryotes arose as a consequence of horizontal gene transfer from eukaryotes.101,103-107 At the molecular level, a study of internal deletions and truncations of E. coli GlnRS has shown that the active site domain of the enzyme, alone, is capable of aminoacylation in vivo. Interestingly, the truncated enzyme exhibits reduced selectivity for aminoacylation of suppressor tRNAs.108 Thus, the Rossmann fold domain may be a prototype of early, broadly-specific enzymes which later diverged to recognize specific tRNA isoacceptors.
Apart from E. coli GlnRS, most studies of GlnRS enzymes distributed throughout Nature have been limited to relatively crude characterization of isolates from natural sources.1 The two major exceptions are the cloning and characterization of the yeast109-111 and human1,74,112-114 genes and proteins. In mammalian cells, GlnRS is a member of a multienzyme complex which includes a total of eight tRNA synthetases together with the three noncatalytic proteins p43, p38 and p18.115,116 Deletion analysis of human GlnRS showed that an N-terminal appended domain comprising 236 amino acids is dispensable to targeting the enzyme for inclusion in the multi-synthetase complex. Instead, the Rossmann fold domain alone suffices for binding to the complex, raising the question of how this active site-containing region can be simultaneously capable of both catalysis and protein-protein interactions.116 Interestingly, however, expression of the GlnRS deletion mutant lacking the N-terminal extension results in exclusion of both ArgRS and p43 from the multienzyme complex. Thus, this region clearly has a role in the protein-protein interactions which build the particle. Further, the N-terminal extension of human GlnRS is also essential to activity: a C-terminal enzyme comprising the entire structure of E. coli GlnRS is inactive when expressed in cell extracts.116 Human GlnRS has also been shown to interact with apoptosis signal-regulating kinase 1 (ASK1) in a glutamine-dependent fashion.117 GlnRS inhibits the apoptosis-inducing activity of ASK1, revealing a new biological role for mammalian tRNA synthetases.
Yeast GlnRS encodes an 809 amino acid protein, with a 225-amino acid N-terminal extension compared with the E. coli enzyme. Unlike the human enzyme, the amino-terminal portion of yeast GlnRS is dispensable for catalytic activity.111,118 This shows that the active site resides, as expected, in the homologous C-terminal region. The role of the appended domain was elucidated upon discovery that E. coli GlnRS is not capable of aminoacylating yeast tRNAGln. This species lacks E. coli tRNAGln identity nucleotides at positions 73 and 3-70 in the acceptor end of the molecule, explaining why cross-species aminoacylation is difficult [wild-type yeast GlnRS similarly fails to aminoacylate E. coli tRNAGln].89 However, a hybrid protein consisting of the yeast appended domain fused to the N-terminus of the E. coli enzyme acquires the ability to aminoacylate yeast tRNA both in vivo and in vitro. The yeast appended domain alone possesses a nonspecific RNA-binding activity, which suggests that it provides additional stabilizing binding interactions to the cross-species E. coli GlnRS-yeast tRNAGln complex.119 Indeed, segments of the nonspecific RNA-binding protein Arc1p fused in the identical position also permit the bacterial enzyme to aminoacylate yeast tRNA.119 The position of the GlnRS N-terminus with respect to tRNA binding suggests that the additional domains in these hybrids may interact with the tRNA core region in the general regions of the D and T-loops. Athough a detailed basis for the new hybrid enzyme activity has not been established, further studies along these lines might yield further insight into pathways of molecular communication inside the complex.
The recent explosion of sequence information across biological domains has resulted in a recent very rapid increase of available glnS sequences. The number of GlnRS sequences is low compared to other tRNA synthetases, since it is absent from so many organisms. Nonetheless, as of this writing 15 sequences have been deposited in databases, mostly as a consequence of whole-genome sequencing efforts. A comprehensive alignment is presented in Figure 9. This shows that GlnRS enzymes possess the greatest degree of similarity in the two halves of the active-site Rossmann fold. The helical subdomain following the fold, which bridges to the anticodon-binding C-terminal domains, also is strongly conserved, while the sequence conservation in the acceptor-binding domain is somewhat less. The anticodon-binding β-barrels are the least well-conserved part of the structure. Although a detailed analysis would be prohibitive in the context of this review, this alignment offers an important resource both for furthering our understanding of structure-function relationships in the enzyme, and for protein engineering efforts.

Acknowledgements
I thank Luke Sherlin for the alignment of GlnRS sequences and for creating Figures 8 and 9. Work in my laboratory related to the topic of this review is supported by NIH grant GM-63713.
References
- 1.
- Freist W, Gauss DH, Ibba M. et al. Glutaminyl-tRNA synthetase. Biol Chem. 1997;378:1103–1117. [PubMed: 9372179]
- 2.
- Ibba M, Hong KW, Söll D. Glutaminyl-tRNA synthetase-from genetics to molecular recognition. Gen Cells. 1996;1:421–427. [PubMed: 9078373]
- 3.
- Englisch-Peters S, Conley J, Plumbridge J. et al. Mutant enzymes and tRNAs as probes of the glutaminyl-tRNA synthetase:tRNAGln interaction. Biochimie. 1991;73:1501–1508. [PubMed: 1725262]
- 4.
- Rogers MJ, Weygand-Durasevic I, Schwob E. et al. Selectivity and specificity in the recognition of tRNA by E coli glutaminyl-tRNA synthetase. Biochimie. 1993;75:1083–1090. [PubMed: 8199243]
- 5.
- Sherman JM, Rogers MJ, Söll D. Recognition in the glutamine tRNA system:from structure to function. In: tRNA: Structure, Biosynthesis and Function, Söll D, Rajbhandary U, eds., (Washington DC: American Society for Microbiology). 1995:395–409.
- 6.
- Rould MA, Steitz TA. Structure of the glutaminyl-tRNA synthetase-tRNAGln-ATP complex. Nucl Acids Mol Biol. 1992;6:225–245.
- 7.
- Rould MA, Perona JJ, Söll D. et al. Structure of glutaminyl-tRNA synthetase complexed with tRNAGln and ATP at 2.8 Å resolution. Science. 1989;246:1135–1142. [PubMed: 2479982]
- 8.
- Rould MA, Perona JJ, Steitz TA. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature. 1991;352:213–218. [PubMed: 1857417]
- 9.
- Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by E. coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. [PubMed: 8364025]
- 10.
- Rath VL, Silvian LF, Beijer B. et al. How glutaminyl-tRNA synthetase selects glutamine. Structure. 1998;6:439–449. [PubMed: 9562563]
- 11.
- McClain WH, Schneider J, Bhattacharya S. et al. The importance of tRNA backbone-mediated interactions with synthetase for aminoacylation. Proc Natl Acad Sci USA. 1998;95:460–465. [PMC free article: PMC18442] [PubMed: 9435214]
- 12.
- Nissan TA, Perona JJ. Alternative designs for construction of the class II transfer RNA tertiary core. RNA. 2000;6:1585–1596. [PMC free article: PMC1370028] [PubMed: 11105758]
- 13.
- Ibba M, Hong K-W, Sherman JM. et al. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc Natl Acad Sci USA. 1996;93:6953–6958. [PMC free article: PMC38915] [PubMed: 8692925]
- 14.
- Hong K-W, Ibba M, Weygand-Durasevic I. et al. Transfer RNA-dependent cognate amino acid recognition by an aminoacyl-tRNA synthetase. EMBO J. 1996;15:1983–1991. [PMC free article: PMC450117] [PubMed: 8617245]
- 15.
- Perona JJ, Swanson R, Steitz TA. et al. Overproduction and purification of E. coli tRNA2Gln and its use in the crystallization of the glutaminyl-tRNA synthetase:tRNAGln complex. J Mol Biol. 1998;202:121–126. [PubMed: 2459391]
- 16.
- Perona JJ, Swanson R, Rould MA. et al. Structural basis for misaminoacylation by mutant E. coli glutaminyl-tRNA synthetase enzymes. Science. 1989;246:1152–1154. [PubMed: 2686030]
- 17.
- Arnez JG, Steitz TA. Crystal structure of unmodified tRNAGln complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure. Biochemistry. 1994;33:7560–7567. [PubMed: 8011621]
- 18.
- Sherlin LD, Bullock TL, Newberry KJ. et al. Influence of transfer RNA tertiary structure on aminoacylation efficiency by glutaminyl and cysteinyl-tRNA synthetases. J Mol Biol. 2000;299:431–446. [PubMed: 10860750]
- 19.
- Bullock TL, Sherlin LD, Perona JJ. Tertiary core rearrangements in a tight-binding transfer RNA aptamer. Nature Struct Biol. 2000;7:497–504. [PubMed: 10881199]
- 20.
- Arnez JG, Steitz TA. Crystal structures of three misacylating mutants of Escherichia coli glutaminyl-tRNA synthetase complexed with tRNAGln and ATP. Biochemistry. 1996;35:14725–14733. [PubMed: 8942633]
- 21.
- Sherlin LD, Bullock TL, Nissan TA. et al. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article: PMC1370207] [PubMed: 11720294]
- 22.
- Hoben P, Royal M, Cheung A. et al. E. coli glutaminyl-tRNA synthetase: characterization of the glnS gene product. J Biol Chem. 1982;257:11644–11650. [PubMed: 6749844]
- 23.
- Yamao F, Inokuchi H, Cheung A. et al. E. coli glutaminyl-tRNA synthetase: isolation and DNA sequence of the glnS gene. J Biol Chem. 1982;257:11639–11643. [PubMed: 6288695]
- 24.
- Uemura H, Conley J, Yamao F. et al. E. coli glutaminyl-tRNA synthetase: a single amino acid replacement relaxes tRNA specificity. Prot Seq Data Anal. 1988a;1:479–485. [PubMed: 2464170]
- 25.
- Rossmann MG, Liljas A, Branden CI. et al. Evolutionary and structural relationships among the dehydrogenases. In: Boyer PD (ed). The Enzymes. Academic Press, NY. 1975;9:61–102.
- 26.
- Perona JJ, Rould MA, Steitz TA. et al. Structural similarities in glutaminyl and methionyl-tRNA synthetases suggest a common overall orientation of tRNA binding. Proc Natl Acad Sci USA. 1991;88:2903–2907. [PMC free article: PMC51348] [PubMed: 2011598]
- 27.
- Rogers MJ, Adachi T, Inokuchi H. et al. Functional communication in the recognition of tRNA by Escherichia coli glutaminyl-tRNA synthetase. Proc Natl Acad Sci USA. 1994;91:291–295. [PMC free article: PMC42933] [PubMed: 7506418]
- 28.
- Das BK, Bhattacharyya T, Roy S. Characterization of a urea induced molten globule intermediate state of glutaminyl-tRNA synthetase from Escherichia coli. Biochemistry. 1995;34:5242–5247. [PubMed: 7711044]
- 29.
- Bhattacharyya A, Mandal AK, Banerjee R. et al. Dynamics of compact denatured states of glutaminyl-tRNA synthetase probed by bis-ANS binding kinetics. Biophys Chem. 2000;87:201–212. [PubMed: 11099182]
- 30.
- Hooper JL, Russell RL, Smith JD. Mischarging in mutant tyrosine transfer RNAs. FEBS Lett. 1972;22:149–155. [PubMed: 11946583]
- 31.
- Shimura Y, Aono H, Ozeki H, et al. Mutant tyrosine tRNA of altered amino acid specificity. FEBS Lett. 1972;22:144–148. [PubMed: 11946582]
- 32.
- Celis JE, Hooper ML, Smith JD. Amino acid acceptor stem of E. coli suppressor tRNATyr is a site of synthetase recognition. Nature New Biol. 1973;244:261–264. [PubMed: 4580701]
- 33.
- Smith JD, Celis JE. Mutant tyrosine transfer RNA that can be charged with glutamine. Nature New Biol. 1973;243:66–71. [PubMed: 4574112]
- 34.
- Ghysen A, Celis JE. Mischarging single and double mutants of Escherichia coli sup3 tyrosine transfer RNA. J Mol Biol. 1974;83:333–351. [PubMed: 4595767]
- 35.
- Inokuchi H, Hoben P, Yamao F. et al. Transfer RNA mischarging mediated by a mutant Escherichia coli glutaminyl-tRNA synthetase. Proc Natl Acad Sci USA. 1984;81:5076–5080. [PMC free article: PMC391640] [PubMed: 6382258]
- 36.
- Uemura H, Rogers MJ, Swanson R. et al. Site-directed mutagenesis to fine-tune enzyme specificity. Protein Engr. 1988b;2:293–296. [PubMed: 3150543]
- 37.
- Hoben P, Uemura H, Yamao F. et al. Misaminoacylation by glutaminyl-tRNA synthetase: relaxed specificity in wild-type and mutant enzymes. Fed Proc. 1984;43:2972. [PubMed: 6389180]
- 38.
- Swanson R, Hoben P, Sumner-Smith M. et al. Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science. 1988;242:1548. [PubMed: 3144042]
- 39.
- Rogers MJ, Söll D. Discrimination between glutaminyl-tRNA synthetase and seryl-tRNA synthetase involves nucleotides in the acceptor stem of tRNA. Proc Natl Acad Sci USA. 1988;85:6627. [PMC free article: PMC282030] [PubMed: 3045821]
- 40.
- Sherman JM, Rogers K, Rogers JM. et al. Synthetase competition and tRNA context determine the in vivo identity of tRNA discriminator mutants. J Mol Biol. 1992a;228:1055–1062. [PubMed: 1474577]
- 41.
- Sherman JM, Rogers MJ, Söll D. Competition of aminoacyl-tRNA synthetases for tRNA ensures the accuracy of aminoacylation. Nucl Acids Res. 1992b;20:2847–2852. [PMC free article: PMC336931] [PubMed: 1377381]
- 42.
- Jahn M, Rogers MJ, Söll D. Anticodon and acceptor stem nucleotides in tRNAGln are major recognition elements for E. coli glutaminyl-tRNA synthetase. Nature. 1991;352:258–260. [PubMed: 1857423]
- 43.
- Nissan TA, Oliphant B, Perona JJ. An engineered class I transfer RNA with a class II tertiary fold. RNA. 1999;5:434–445. [PMC free article: PMC1369771] [PubMed: 10094311]
- 44.
- Weygand-Durasevic I, Schwob E, Söll D. Acceptor-end binding domain interactions ensure correct aminoacylation of transfer RNA. Proc Natl Acad Sci USA. 1993;90:2010–2014. [PMC free article: PMC46010] [PubMed: 7680483]
- 45.
- Sherman JM, Söll D. Aminoacyl-tRNA synthetases optimize both cognate tRNA recognition and discrimination against noncognate tRNAs. Biochemistry. 1996;35:601–607. [PubMed: 8555233]
- 46.
- Hayase Y, Jahn M, Rogers MJ. et al. Recognition of bases in Escherichia coli tRNAGln by glutaminyl-tRNA synthetase: a complete identity set. EMBO J. 1992;11:4159–4165. [PMC free article: PMC556926] [PubMed: 1396597]
- 47.
- Sherman JM, Thomann H-U, Söll D. Functional connectivity between tRNA binding domains in glutaminyl-tRNA synthetase. J Mol Biol. 1996;256:818–828. [PubMed: 8601833]
- 48.
- Yaniv M, Folk WR, Berg P. et al. A single mutational modification of a tryptophan-specific transfer RNA permits aminoacylation by glutamine and translation of the codon UAG. J Mol Biol. 1974;86:245–260. [PubMed: 4606150]
- 49.
- Celis JE, Coulondre C, Miller JH. Suppressor su+7 inserts tryptophan in addition to glutamine. J Mol Biol. 1976;104:729–734. [PubMed: 781298]
- 50.
- Yarus M, Knowlton R, Söll L. 1977.Aminoacylation of the ambivalent su+7 amber suppressor tRNA In: Nucleic acid - Protein Rcognition (HJ Vogel, ed.), Academic Press, New York, p. 391 .
- 51.
- Rogers MJ, Adachi T, Inokuchi H. et al. Switching tRNAGln identity from glutamine to tryptophan. Proc Natl Acad Sci USA. 1992;89:3463–3467. [PMC free article: PMC48888] [PubMed: 1565639]
- 52.
- Schulman LH, Pelka H. In vitro conversion of a methionine to glutamine-acceptor tRNA. Biochemistry. 1985;24:7309–7314. [PubMed: 3910101]
- 53.
- Seong BL, Lee C-P, RajBhandary UL. Suppression of amber codons in vivo as evidence that mutants derived from Escherichia coli initiator tRNA can act at the step of elongation in protein synthesis. J Biol Chem. 1989;264:6504–6508. [PubMed: 2649502]
- 54.
- Varshney U RajBhandaryUL. Initiation of protein synthesis from a termination codon. Proc Natl Acad Sci USA. 1990;87:1586–1590. [PMC free article: PMC53520] [PubMed: 2406724]
- 55.
- Varshney U, Lee C-P, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 1991;266:24712–24718. [PubMed: 1761566]
- 56.
- Tsai F, Curran JF. tRNA2Gln mutants that translate the CGA arginine codon as glutamine in Escherichia coli. RNA. 1998;4:1514–1522. [PMC free article: PMC1369722] [PubMed: 9848650]
- 57.
- Robertus JD, Ladner JE, Finch JT. et al. Structure of yeast tRNAPhe at 3 Å resolution. Nature. 1974;250:546. [PubMed: 4602655]
- 58.
- Moras D, Comarmond MB, Fischer J, et al. Crystal structure of yeast tRNAAsp. Nature. 1980;288:669. [PubMed: 7005687]
- 59.
- Urbonavicius J, Qian Q, Durand JMB. et al. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001;20:4863–4873. [PMC free article: PMC125605] [PubMed: 11532950]
- 60.
- Seno T, Agris PF, Söll D. Involvement of the anticodon region of Escherichia coli tRNAGln and tRNAGlu in the specific interaction with cognate aminoacyl-tRNA synthetase. Alteration of the 2-thiouridine derivatives located in the anticodon of the tRNAs by BrCN or sulfur deprivation. Biochim Biophys Acta. 1974;349:328–338. [PubMed: 4366808]
- 61.
- Rogers KC, Söll D. Discrimination among tRNAs intermediate in glutamate and glutamine acceptor identity. Biochemistry. 1993;32:14210–14219. [PubMed: 7505112]
- 61a.
- Rogers KC, Crescenzo AT, Söll D. Aminoacylation of transfer RNAs with 2-thiouridine derivatives in the wobble position of the anticodon. Biochimie. 1995;77:66–74. [PubMed: 7541255]
- 62.
- Frugier M, Söll D, Giege R. et al. Identity switches between tRNAs aminoacylated by class I glutaminyl- and class II aspartyl-tRNA synthetases. Biochemistry. 1994;33:9912–9921. [PubMed: 8060999]
- 63.
- McClain WH, Seidman JG. Genetic conversion of G-C base-pairs to A-U base-pairs in a transfer RNA. J Mol Biol. 1987;197:605–608. [PubMed: 3441013]
- 64.
- McClain WH, Schneider J, Gabriel K. Association of tRNAGln acceptor identity with phosphate-sugar backbone interactions observed in the crystal structure of the Escherichia coli glutaminyl-tRNA synthetase-tRNAGln complex. Biochimie. 1993;75:1125–1136. [PubMed: 8199248]
- 65.
- Buttcher V, Senger B, Schumacher S. et al. Modulation of the suppression efficiency and amino acid identity of an artificial yeast amber isoleucine transfer RNA in Escherichia coli by a G-U pair in the anticodon stem. Biochem Biophys Res Commun. 1994;200:370–377. [PubMed: 8166708]
- 66.
- Stubenrauch M. In vitro selection and characterization of tRNA-like substrates of E. coli glutaminyl-tRNA synthetase. Doctoral thesis, Dept. of Chemistry & Biochemistry, University of Colorado, Boulder CO. (1996).
- 67.
- Brick P, Bhat TN, Blow DM. Structure of tyrosyl-tRNA synthetase refined at 2.3 Å J Mol Biol. 1989;resolution interaction of the enzyme with the tyrosyl adenylate intermediate208:83–98. [PubMed: 2504923]
- 68.
- Bernier S, Dubois DY, Therrien M. et al. Synthesis of glutaminyl adenylate analogues that are inhibitors of glutaminyl-tRNA synthetase. Bioorg Med Chem Lett. 2000;10:2441–2444. [PubMed: 11078196]
- 69.
- Schon A, Hottinger H, Söll D. Protein biosynthesis in organelles requires misaminoacylation of tRNA. Nature. 1988;331:187–190. [PubMed: 3340166]
- 70.
- Curnow AW, Hong K, Yuan R. et al. Glu-tRNAGln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci USA. 1997;94:11819–11826. [PMC free article: PMC23611] [PubMed: 9342321]
- 71.
- Ibba M, Becker HD, Stathopoulos C. et al. The adaptor hypothesis revisited. Trends in Biochem Sci. 2000;25:311–316. [PubMed: 10871880]
- 72.
- Liu J, Ibba M, Hong K-W, Söll D. The terminal adenosine of tRNAGln mediates tRNA-dependent amino acid recognition by glutaminyl-tRNA synthetase. Biochemistry. 1998;37:9836–9842. [PubMed: 9657697]
- 73.
- Hong K-W, Ibba M, Söll D. Retracing the evolution of amino acid specificity in glutaminyl-tRNA synthetase. FEBS Lett. 1998;434:149–154. [PubMed: 9738468]
- 74.
- Agou F, Quevillon S, Kerjan P. et al. Switching the amino acid specificity of an aminoacyl-tRNA synthetase. Biochemistry. 1998;37:11309–11314. [PubMed: 9698378]
- 75.
- Liu DR, Schultz PG. Progress toward the evolution of an organism with an expanded genetic code. Proc Natl Acad Sci USA. 1999;96:4780–4785. [PMC free article: PMC21768] [PubMed: 10220370]
- 76.
- Kowal AK, Kohrer C, RajBhandary UL. Twenty-first aminoacyl-tRNA synthetase-suppressor tRNA pairs for possible use in site-specific incorporation of amino acid analogues into proteins in eukaryotes and in eubacteria. Proc Natl Acad Sci USA. 2001;98:2268–2273. [PMC free article: PMC30127] [PubMed: 11226228]
- 77.
- Liu DR, Magliery TJ, Pastrnak M. et al. Engineering a tRNA and aminoacyl-tRNA synthetase for the site-specific incorporation of unnatural amino acids into proteins in vivo. Proc Natl Acad Sci USA. 1997;94:10092–10097. [PMC free article: PMC23315] [PubMed: 9294168]
- 78.
- Kern D, Potier S, Lapointe J. et al. The glutaminyl-tRNA synthetase of Escherichia coli. Purification, structure and function relationship. Biochim Biophys Acta. 1980;607:65–80. [PubMed: 6989402]
- 79.
- Fersht AR. Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry. 1987;26:8031–8037. [PubMed: 3442641]
- 80.
- Fersht AR, Knill-Jones JW, Bedouelle H. et al. Reconstruction by site-directed mutagenesis of the transition state for the activation of tyrosine by the tyrosyl-tRNA synthetase:A mobile loop envelopes the transition state in an induced-fit mechanism. Biochemistry. 1988;27:1581–1587. [PubMed: 3284584]
- 81.
- Kern D, Lapointe J. The twenty aminoacyl-tRNA synthetases from Escherichia coli. General separation procedure, and comparison of the influence of pH and divalent cations on their catalytic activities. Biochimie. 1979;61:1257–1272. [PubMed: 44203]
- 82.
- Garcia GA, Leatherbarrow RJ, Eckstein F. et al. Metal ion dependence of phosphorothioate ATP analogues in the Bacillus stearothermophilus tyrosyl-tRNA synthetase reaction. Biochemistry. 1990;29:1643–1648. [PubMed: 2334722]
- 83.
- Connolly BA, Von der Haar F, Eckstein F. Structure of the metal.nucleotide complex in the yeast phenylalanyl transfer ribonucleic acid synthetase reaction as determined with diastereomeric phosphorothioate analogs of ATP. J Biol Chem. 1980;255:11301–11307. [PubMed: 7002919]
- 84.
- Smith LT, Cohn M. Reactions of thio analogues of adenosine 5'-triphosphate catalyzed by methionyl-tRNA synthetase from Escherichia coli and metal dependence of stereospecificity. Biochemistry. 1982;21:1530–1534. [PubMed: 7044416]
- 85.
- Wada M, Sekine K, Itikawa H. Participation of the dnaK and dnaJ gene products in phosphorylation of glutaminyl-tRNA synthetase and threonyl-tRNA synthetase of E. coli K12. J Bacteriol. 1986;168:213–220. [PMC free article: PMC213440] [PubMed: 3531168]
- 86.
- Itikawa H, Wada M, Sekine K. et al. Phosphorylation of glutaminyl-tRNA synthetase and threonyl-tRNA synthetase by the gene products of dnaK and dnaJ in Escherichia coli K-12 cells. Biochimie. 1989;71:1079–1087. [PubMed: 2512999]
- 87.
- Pendergast AM, Venema RC, Traugh JA. Regulation of phosphorylation of aminoacyl-tRNA synthetases in the high molecular weight core complex in reticulocytes. J Biol Chem. 1987;262:5939–5942. [PubMed: 2437110]
- 88.
- Thomann HU, Ibba M, Kong -K-W. et al. Homologous expression and purification of mutants of an essential protein by reverse epitope-tagging. Bio-Technology. 1996;14:50–55. [PubMed: 9636312]
- 89.
- Whelihan EF, Schimmel P. Rescuing an essential enzyme-RNA complex with a non-essential appended domain. EMBO J. 1997;16:2968–2974. [PMC free article: PMC1169904] [PubMed: 9184240]
- 90.
- Bhattacharyya T, Bhattacharyya A, Roy S. A fluorescence spectroscopic study of glutaminyl-tRNA synthetase from Escherichia coli and its implications for the enzyme mechanism. Eur J Biochem. 1991;200:739–745. [PubMed: 1915346]
- 91.
- Bhattacharyya T, Roy S. A fluorescence spectroscopic study of substrate-induced conformational changes in glutaminyl-tRNA synthetase. Biochemistry. 1993;32:9268–9273. [PubMed: 8369295]
- 92.
- Hoben P, Söll D. Glutaminyl-tRNA synthetase of Escherichia coli. Meth Enzymol. 1985;113:55–59. [PubMed: 3911010]
- 93.
- Lloyd AJ, Thomann H-U, Ibba M. et al. A broadly applicable continuous spectrophotometric assay for measuring aminoacyl-tRNA synthetase activity. Nucl Acids Res. 1995;23:2886–2892. [PMC free article: PMC307126] [PubMed: 7659511]
- 94.
- Wolfson AD, Pleiss JA, Uhlenbeck OC. A new assay for tRNA aminoacylation kinetics. RNA. 1998;4:1019–1023. [PMC free article: PMC1369678] [PubMed: 9701292]
- 95.
- Perona JJ. Crystal structure of the E. coli glutaminyl-tRNA synthetase:tRNAGln complex. Doctoral thesis, Department of Molecular Biophysics & Biochemistry, Yale University. 1990
- 96.
- Mandal AK, Bhattacharyya A, Bhattacharyya S. et al. A cognate tRNA specific conformational change in glutaminyl-tRNA synthetase and its implication for specificity. Protein Science. 1998;7:1046–1051. [PMC free article: PMC2143984] [PubMed: 9568911]
- 97.
- Beresten S, Jahn M, Söll D. Aminoacyl-tRNA synthetase-induced cleavage of tRNA. Nucleic Acids Res. 1992;20:1523–1530. [PMC free article: PMC312233] [PubMed: 1579445]
- 98.
- Kitabatake M, Ibba M, Hong K-W. et al. Genetic analysis of functional connectivity between substrate recognition domains of Escherichia coli glutaminyl-tRNA synthetase. Mol Gen Genet. 1996;252:717–722. [PubMed: 8917315]
- 99.
- Weygand-Durasevic I, Rogers MJ, Söll D. Connecting anticodon recognition with the active site of Escherichia coli glutaminyl-tRNA synthetase. J Mol Biol. 1994;240:111–118. [PubMed: 8027995]
- 99a.
- Wright DJ, Martinis SA, Jahn M. et al. Acceptor stem and anticodon RNA hairpin helix interactions with glutamine tRNA synthetase. Biochimie. 1993;75:1041–1049. [PubMed: 8199240]
- 100.
- Bahar I, Jernigan RL. Vibrational dynamics of transfer RNAs: Comparison of the free and synthetase-bound forms. J Mol Biol. 1998;281:871–884. [PubMed: 9719641]
- 101.
- Woese CR, Olsen GJ, Ibba M. et al. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–236. [PMC free article: PMC98992] [PubMed: 10704480]
- 102.
- Nabholz CE, Hauser R, Schneider A. Leishmania tarentolae contains distinct cytosolic and mitochondrial glutaminyl-tRNA synthetase activities. Proc Natl Acad Sci USA. 1997;94:7903–7908. [PMC free article: PMC21527] [PubMed: 9223285]
- 103.
- Di GiulioM. Origin of glutaminyl-tRNA synthetase: an example of palimpsest? J Mol Evol. 1993;37:5–10. [PubMed: 8360919]
- 104.
- Rogers KC, Söll D. Divergence of glutamate and glutamine aminoacylation pathways: providing the evolutionary rationale for mischarging. J Mol Evol. 1995;40:476–481. [PubMed: 7783222]
- 105.
- Siatecka M, Rozek M, Barciszewski J. et al. Modular evolution of the Glx-tRNA synthetase family — rooting of the evolutionary tree between the bacteria and archaea eukarya branches. Eur J Biochem. 1998;256:80–87. [PubMed: 9746349]
- 106.
- Brown JR, Doolittle WF. Gene descent, duplication, and horizontal transfer in the evolution of glutamyl- and glutaminyl-tRNA synthetases. J Mol Evol. 1999;49:485–495. [PubMed: 10486006]
- 107.
- Handy J, Doolittle RF. An attempt to pinpoint the phylogenetic introduction of glutaminyl-tRNA synthetase among bacteria. J Mol Evol. 1999;49:709–715. [PubMed: 10594171]
- 108.
- Schwob E, Söll D. Selection of a ‘minimal’ glutaminyl-tRNA synthetase and the evolution of class I synthetases. EMBO J. 1993;12:5201–5208. [PMC free article: PMC413784] [PubMed: 7505222]
- 109.
- Ludmerer SW, Schimmel P. Cloning of GLN4: an essential gene that encodes glutaminyl-tRNA synthetase in Saccharomyces cerevisiae. J Bacteriol. 1985;163:763–768. [PMC free article: PMC219187] [PubMed: 2991203]
- 110.
- Ludmerer SW, Schimmel P. Gene for yeast glutamine tRNA synthetase encodes a large amino-terminal extension and provides a strong confirmation of the signature sequence for a group of the aminoacyl-tRNA synthetases. J Biol Chem. 1987a;262:10801–10806. [PubMed: 3301841]
- 111.
- Ludmerer SW, Schimmel P. Construction and analysis of deletions in the amino-terminal extension of glutaminyl-tRNA synthetase of Saccharomyces cerevisiae. J Biol Chem. 1987b;262:10807–10813. [PubMed: 3301842]
- 112.
- Thommes P, Fett R, Schray B. et al. The core region of human glutaminyl-tRNA synthetase homologies with the Escherichia coli and yeast enzymes. Nucl Acids Res. 1988;16:5391–5406. [PMC free article: PMC336774] [PubMed: 3290852]
- 113.
- Fett R, Knippers R. The primary structure of human glutaminyl-tRNA synthetase. A highly conserved core, amino acid repeat regions, and homologies with translation elongation factors. J Biol Chem. 1991;266:1448–1455. [PubMed: 1988429]
- 114.
- Kaiser E, Eberhard D, Knippers R. Exons encoding the highly conserved part of human glutaminyl-tRNA synthetase. J Mol Evol. 1992;34:45–53. [PubMed: 1556743]
- 115.
- Schray B, Thommes P, Knippers R. Glutaminyl-tRNA synthetase as a component of the high-molecular weight complex of human aminoacyl-tRNA synthetases. An immunological study. Biochim Biophys Acta. 1990;1087:226–234. [PubMed: 2223884]
- 116.
- Kim T, Park SG, Kim JE. et al. Catalytic peptide of human glutaminyl-tRNA synthetase is essential for its assembly to the aminoacyl-tRNA synthetase complex. J Biol Chem. 2000;275:21768–21772. [PubMed: 10801842]
- 117.
- Ko Y-G, Kim E-K, Kim T. et al. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:6030–6036. [PubMed: 11096076]
- 118.
- Ludmerer SW, Wright DJ, Schimmel P. Purification of glutamine tRNA synthetase from Saccharomyces cerevisiae. A monomeric aminoacyl-tRNA synthetase with a large and dispensable NH2-terminal domain. J Biol Chem. 1993;268:5519–5523. [PubMed: 8449914]
- 119.
- Wang C-C, Schimmel P. Species barrier to RNA recognition overcome with nonspecific RNA binding domains. J Biol Chem. 1999;274:16508–16512. [PubMed: 10347214]
- 120.
- Bullock T, Uter N, Nissan TA, Perona JJ. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol. 2003;328:395–408. [PubMed: 12691748]
- 121.
- Sherlin LD, Perona JJ. tRNA-dependent active site assembly in a class I aminoacyl-tRNA synthetase. Structure. 2003;11:591–603. [PubMed: 12737824]
- 122.
- Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci USA. 2002;99:5965–5970. [PMC free article: PMC122885] [PubMed: 11983895]
- Glutaminyl-tRNA Synthetases - Madame Curie Bioscience DatabaseGlutaminyl-tRNA Synthetases - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...