NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Arvin A, Campadelli-Fiume G, Mocarski E, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007.
Introduction
Herpes simplex virus (HSV) represents the most comprehensive example of virus-receptor interaction in the Herpesviridae family, and the prototype virus encoding multipartite entry genes. Whereas small enveloped viruses package the functions required for entry and fusion into one or two fusion glycoproteins, in HSV the same functions are distributed over several distinct glycoproteins, each with a specialized activity. In addition, HSV encodes a highly sophisticated system for promoting and blocking fusion between the viral envelope and cell membrane. Because the most obvious models of virus entry into the cell do not fit with the HSV complexity, and despite our detailed knowledge of the HSV receptors and of the crystal structure of glycoprotein D (gD), the receptor-binding glycoprotein, and of gB, HSV entry is still, in part, a puzzle (WuDunn and Spear, 1989; Cocchi et al., 1998b; Geraghty et al., 1998; Carfi et al., 2001).
The current model of HSV entry envisions that, first, the virus attaches to cell membranes by the interaction of gC, and possibly gB, to glycosaminoglycans (GAGs) (Herold et al., 1991). This binding likely creates multiple points of adhesion, is reversible, and the detached virus maintains its infectivity, indicating that fusion has yet to take place. Penetration requires gD, whose ectodomain contains two physically separate and functionally distinct regions, i.e., the region made of the N-terminus that carries the receptor-binding sites, and the C-terminus that carries the profusion domain (Ligas and Johnson, 1988; Cocchi et al., 2004). The role of gD in entry is to interact with one of the entry receptors, to signal receptor-recognition and thus trigger fusion, by recruiting three additional glycoproteins – gB, gH, gL. The trio of gB, gH, gL, execute fusion with the plasma membrane or endocytic vesicle of the target cell (Fig. 7.1) (Cai, W. H. et al., 1988; Forrester et al., 1992; Roop et al., 1993; Campadelli-Fiume et al., 2000; Spear et al., 2000; Nicola et al., 2003). Of these, gH carries elements characteristic of viral fusion glycoproteins, i.e., a hydrophobic α-helix with attributes of an internal fusion peptide, and two heptad repeats with propensity to form coiled coils (Gianni et al., 2005a,b). gB is a trimer with a coiled coil core; its structure closely resembles that of viral fusion proteins (Heldwein et al., 2006; Roche et al., 2006). Following fusion, the released tegumented nucleocapsid travels along microtubules to the nuclear pore, where the viral DNA is released into the nucleus (Sodeik et al., 1997).
Much less is known about varicella zoster virus (VZV) entry. The process may be very different from that of HSV inasmuch as virion-to-cell infection is inefficient in the VZV system, and the viral genome presents a striking difference from that of HSV, namely the lack of gD (Davison and Scott, 1986).
The membrane proteins
The HSV envelope contains at least eleven glycoproteins (gB, gC, gD, gE, gG, gH, gI, gJ, gK, gL, and gM). Additional membrane proteins not detected in the extracellular virion envelope are UL 20, UL 34, UL 45 and possibly US 9. The transcripts for UL 24, UL 43, and UL 49.5 ORFs have been recognized, but the proteins have yet to be identified. A summary list with references is presented in Chapter 6.
From a structural point of view, the majority of HSV glycoproteins are type Ⅰ glycoproteins. Variants include gL, which is soluble (Hutchinson et al., 1992a); gB, which may carry two or three α-helices in the transmembrane (TM) region (Pellett et al., 1985) and gK, gM, and UL 20, which carry information for multiple transmembrane segments (McGeoch et al., 1988). US 9 and HSV -2 UL 45 are type Ⅱ glycoproteins with a C-terminal ectodomain.
At the ultrastructural level, HSV glycoproteins form long thin spikes, each made of a single species. As visualized in cryo-electron tomograms of isolated virions, the envelope contains 600–750 glycoprotein spikes that vary in length, spacings, and in the angle at which they emerge from the membrane. Their distribution in the envelope suggests functional clustering (Grunewald et al., 2003). In contrast, slender spikes have not been seen in varicella zoster virions (VZV) grown in cultured cells. Instead, the virion appears to be covered by an envelope studded with protrusions rather than spikes. An example is shown in Fig. 7.2. Further studies at even higher resolution will be required to determine differences between HSV and VZV envelopes.
HSV
Attachment to cells
Attachment of HSV to cells occurs upon binding of gC to GAGs that decorate heparan sulphate or chondroitin sulphate (Spear et al., 1992) (Fig. 7.1). This step enhances HSV infectivity, but is not an absolute requirement, as cells defective in heparan sulphate and chondroitin sulphate exhibit a 100-fold reduced susceptibility to infection, yet can be infected (Gruenheid et al., 1993). A large variety of viruses use heparan sulphate proteoglycans as receptors; their broad expression argues that they can not be responsible for any specific viral tropism.
The major actor during attachment is gC, a non essential glycoprotein encoded by the UL 44 gene. gC is a mucin-type glycoprotein because of its high content in N-linked and O-linked oligosaccharides. Its ectodomain structure is provided in part by 8 cysteines, and harbors two physically separate antigenic regions, antigenic sites Ⅰ and Ⅱ, that map at the C- and N-termini of the molecule, respectively (Dolter et al., 1992). Evidence for the role of gC in attachment rests on several lines of evidence. Initially, it was observed that the polycations neomycin and polylysine inhibit attachment of HSV -1, but not HSV -2 to cells, and this differential effect was mapped to gC (Campadelli-Fiume et al., 1990). gC and gB bind heparin-Sepharose columns (Herold et al., 1991). The affinity of binding to heparan sulphate is on the order of 10−8 M (Rux et al., 2002). The region important for the interaction with heparan sulphate maps to the N-terminus of gC (Tal-Singer et al., 1995). Virion binding to cells is reduced in HSV -1 mutants lacking gC-type1 gene, but not in HSV -2 mutants lacking gC-type2 (Herold et al., 1991; Gerber et al., 1995). The majority of these studies were performed with mutants constructed in the background of the HSV -1(KOS) strain. In contrast, deletion of gC gene in the genetic background of Sc16 and HFEM strains yielded viruses with unimpaired attachment activity, suggesting that in different virus strains, attachment may be carried out by different proteins (Griffiths et al., 1998). gC has also been implicated in virus attachment to the baso-lateral domain of MDCK polarized epithelial cells, and to the apical domain of polarized human CaCo2 cells.
Interaction of gD with its receptors
gD
The ectodomain of gD is required and sufficient to enable HSV entry into cells. It is made of two separate and distinct regions, i.e., the N-terminus, carrying the receptor-binding sites (approximately contained between residues 1 and 250–260), and the C-terminus carrying the profusion domain (residues 250–260 to 305) (Cocchi et al., 2004).
A breakthrough in our understanding of HSV entry came from resolution of the crystal structure of a soluble form of gD, initially up to amino acid residue 259 (Carfi et al., 2001), and later on up to residue 306 (Krummenacher et al., 2005). The initial structure was determined for gD alone and for gD in complex with one of the gD receptors, HVEM (herpesvirus entry mediator) (Fig. 7.3). The N-terminus consists of three portions, an Ig-folded central core (residues 56–184) made of β-strands forming two antiparallel β-sheets, and two extensions, one N-terminal (residues 1–37) and one C-terminal (Fig. 7.3). The N-terminus, which harbors all the contact residues to HVEM, is disordered in the crystal of gD alone, but forms a hairpin when gD is complexed with HVEM. gD and HVEM thereby form an intermolecular β-sheet, which is believed to stabilize the complex. Formation of the N-terminal hairpin documents a conformational change to gD when it binds HVEM .
The crystal structure of gD in complex with nectin1 has not yet been solved. The nectin1-binding site on gD, determined by means of insertion-deletion or substitution mutants, appears to be more widespread than the HVEM interaction region, (Milne et al., 2003; Yoon et al., 2003; Zhou et al., 2003; Jogger et al., 2004; Manoj et al., 2004; Connolly et al., 2005). The only recombinant described so far debilitated for interaction with nectin1 carries the V34S substitution (Zhou and Roizman, 2006).
Receptors
Entry receptors interact with gD. This notion was established long before the actual receptors were identified, and rests on two lines of evidence. First, soluble gD binds in a saturable manner to cells and prevents infection (Johnson, D. C. et al., 1990; Nicola et al., 1997). Second, expression of gD from a transgene renders cells resistant to infection, because of its ability to sequester the receptor, a phenomenon designated restriction to infection or gD-mediated interference (Campadelli-Fiume et al., 1988; Johnson, R. M. and Spear, 1989).
The search for HSV receptors was an active field in the 1990s, and a number of molecules were described as potential receptors. A breakthrough came from Spear and coworkers, who made use of HSV -resistant CHO cells, and identified a HeLa cell cDNA clone that encoded HVEM (Montgomery et al., 1996). However, three observations in that study suggested the existence of additional receptors. First, HVEM appeared to be expressed by a limited number of cell lines. Second, antibodies to HVEM failed to completely block HSV infection. Third, several virus strains were unable to enter CHO cells expressing HVEM but were otherwise viable. This boosted further efforts in the field, which quickly led to the discovery of nectins, and, later on, of modified heparan sulphate. Altogether, the receptors known to date belong to three unrelated molecular families. Their present and past nomenclature, and the viruses for which they serve as receptors are reported in the table.
Nectins are intercellular adhesion molecules
Research in the field of nectins has proceeded in parallel with their characterization as HSV receptors (Takai et al., 2003). Nectins 1–4 form a subfamily of Ca2+-independent immunoglobulin (Ig)-type intercellular adhesion molecules. Together with nectin-like molecules and poliovirus receptor, they share the same overall structure consisting of three Ig-type domains. Splice variant isoforms are designated with Greek letters. Nectins form homo cis-dimers on the plasma membranes and trans-dimers with nectins present on the adjacent cell. Each nectin has a specialized pattern of trans-dimer formation with itself or other nectins (Reymond et al., 2000; Takai et al., 2003). Their main attribute is the formation, together with cadherins, of the adherens junctions of epithelial cells, and in cooperation or not with cadherins, the organization of claudin-based tight junctions. In addition, they are involved in the formation of synapses in neurons and the organization of heterotypic junctions between Sertoli cells and spermatids in the testis (Takai et al., 2003).
Most nectins carry a C-terminal conserved motif that binds afadin, thus anchoring the adhesion molecules to the cytoskeleton (Takai et al., 2003). This domain is absent from nectin1β which is not restricted to adherens junctions. Nectin-mediated signalling activity leads to activation of a variety of extracellular and intracellular molecules, such as scatter factor/hepatocyte growth factor, Ras, Cdc42 and Rac small G proteins (Takai et al., 2003).
Nectin1
Human nectin1 is a broad spectrum receptor for human and animal alphaherpesviruses (Table 7.1). Three isoforms are known, two of which: -α and -β are membrane-bound (Cocchi et al., 1998a,b; Geraghty et al., 1998; Krummenacher et al., 1998; Krummenacher et al., 1999; Campadelli-Fiume et al., 2000). Their main properties as HSV receptors are as follows.
(ⅰ) Nectin1 is broadly expressed in human tissues, including tissues and organs targeted by HSV, like CNS, ganglia and muco-epithelia (Cocchi et al., 1998b; Haarr et al., 2001; Matsushima et al., 2003; Richart et al., 2003; Linehan et al., 2004). It is expressed in virtually all human cell lines, including epithelial cells, neurons and fibroblasts (Campadelli-Fiume et al., 2000; Spear et al., 2000). Some of these cells simultaneously express HVEM (Krummenacher et al., 2004).
When HSV initially infects mucosal epithelium, the apical domain of polarized epithelial cells are targeted initially, whereas basolateral domains of epithelial cells are available to the virus only if a lesion disrupts the integrity of the lining. It was therefore of interest to know whether nectin1 can serve as an HSV receptor in polarized epithelial cells. Human CaCo2 cells can be infected with HSV from the apical domain (Griffiths et al., 1998), whereas MDCK cells and primary human keratinocytes are preferentially infected from the basolateral domain (Schelhaas et al., 2003; Marozin et al., 2004). These differences may reflect cell line-dependent differences in the pattern of polarization of a same molecule, or distribution of receptors like nectin1-β and HVEM, which do not appear to be restricted to adherens junctions.
(ⅱ) Nectin1 interacts physically with gD (Krummenacher et al., 1999). The interaction requires the first 250 residues of gD and the V domain of nectin1. The affinity ranges from 10−6 to 10−8 molar, with the highest affinity observed with forms of gD that were truncated at or after residue 250. Affinity decreases by 100-fold with gD306t, reflecting a folding of the most C-terminal portion of gD towards the core (Whitbeck et al., 1999; Krummenacher et al., 2005). Insertion mutations alter the binding affinity; insertions at the N-terminus (e.g., at residues 34, and 43) modify the binding to HVEM but not to nectin1 (Milne et al., 2003; Jogger et al., 2004). Remarkably, even when the binding affinities are low, or undetectable, the mutant forms of gD maintain the ability to mediate HSV entry and cell–cell fusion, implying that gD functions in virus entry and cell fusion regardless of its receptor-binding affinity and kinetics, and that as long as interaction with a functional receptor occurs, entry takes place (Milne et al., 2003; Zhou et al., 2003).
Human nectin1 also binds isoforms of gD from animal α-herpesviruses. The affinity may be even higher than for HSV -1 gD (in the case of PrV), or very low (in the case of BHV -1) (Connolly et al., 2001). Even when the affinity is very low, human nectin1 is capable of mediating entry (Cocchi et al., 1998b; Geraghty et al., 1998).
(ⅲ) The domain of nectin1 functional in HSV entry and in binding to gD was initially mapped to the N-terminal V domain, and subsequently to the C-C′-C′′ ridge (Krummenacher et al., 2000; Cocchi et al., 2001; Menotti et al., 2002b). Critical residues that may be part of the interface with gD are amino acids 77 and 85 (Martinez and Spear, 2002).
(ⅳ) Nectin1-γ is a natural soluble isoform of nectin1 generated by alternative splicing. Although it contains the three Ig domains, it has a narrow distribution in human tissues, unlike nectin1-α and -β. Like soluble recombinant forms of nectin1, it has the capacity to bind to virions and block infectivity. An unexpected property was that the soluble nectin1-γ molecules suffices to mediate virus entry into receptor-negative cells. This may be consequent either to an association to endogenous nectins, or to a direct binding of the soluble receptor to virions (Lopez et al., 2001).
Nectin 2 and the unrestricted or rid mutations
The remarkable feature about nectin2 is that a single amino acid substitution in gD confers to HSV the ability to use nectin2 as an alternative receptor, without hampering its ability to use nectin1 (Table 7.1). At the same time, this mutation abolishes the interaction with HVEM (Connolly et al., 2003; Yoon et al., 2003). The end result is that the host range of the virus is modified. The mutations are L25P, Q27P, or Q27R, and are present in the unrestricted, or rid HSV -1 mutants. Nectin2 also serves as a weak receptor for some strains of HSV -2, but is inactive for wt-HSV-1 (Warner et al., 1998; Lopez et al., 2000; Krummenacher et al., 2004). Physical interaction studies were in agreement with these properties (Warner et al., 1998; Lopez et al., 2000; Yoon et al., 2003). The nectin2 residues critical for HSV entry were identified as amino acids 75–81 and 89, which lie adjacent to the predicted C′C′′ β-strands, i.e., the region corresponding to the nectin1 region involved in interaction with wild-type gD.
The TNF receptor family
TNFRs (tumor necrosis factor receptors) form a family of signal transduction molecules involved in regulation of cell proliferation, differentiation and apoptotic death. Structurally, their ectodomain is composed of four typical cysteine-rich domains (CRDs). The family includes twenty nine human members, classified into three groups according to their cytoplasmic sequences and signaling properties. Members of the first group (exemplified by Fas) contain a death domain (DD) in the cytoplasmic tail. After binding to their ligands they interact with intracellular adaptors, which, in turn, induce apoptosis by activation of the caspase cascade. Members of the second group (exemplified by TNFR 2 and HVEM) lack a death domain, and instead contain one or more TRAF (TNFR-associated factor) interacting motifs (TIMs), which trigger a variety of signal transduction pathways, including those for activation of nuclear factor κB (NF-κB), Jun N-terminal kinase (JNK), p38, extracellular signal-related kinase (ERK) and phosphoinositide 3-kinase (PI3K). Members of the third group (e.g., TNFR 3, TNFR 4, etc.) lack intracellular signaling motifs, and act as decoy receptors.
The natural ligands of the TNFRs are a family of cytokines whose prototype is TNF, and include lymphotoxin (LT)α, LT β, and LIGHT. They are biologically active as trimers: their binding to the receptors causes the trimerization of the intracellular domains, which, in turn, interact with high affinity with trimeric cellular adaptors (e.g., TRAF s). Each cytokine interacts with more than one receptor.
HVEM
HVEM was first identified as a HSV receptor and was classified as a novel member of the TNFR family based on structural motifs (Montgomery et al., 1996). The cytoplasmic tail interacts with several members of the TRAF family, leading to the activation of targets like NF -κB, Jun N-terminal kinase, and AP -1, and the consequent induction of T cell activation, proliferation, cytokine release, and expression of cell surface activation markers (Harrop et al., 1998). Its ligands are LT α3 and LIGHT (Mauri et al., 1998). LIGHT –HVEM interactions contribute to the cytotoxic T-lymphocyte-mediated immune response.
The main properties of HVEM as an HSV receptor are as follows.
- HVEM binds wild-type gD. The affinity of the binding is of the same order of magnitude as that of nectin1/gD binding, and the interaction requires the same region of gD, i.e., the first 250 residues, or longer (Willis et al., 1998). gD and LIGHT compete with each other for the binding to HVEM; accordingly, LIGHT interferes with HSV entry in HVEM -expressing cells (Mauri et al., 1998).
- The gD contact site on HVEM involves CRD 1 and 2, with the majority of contacts lying in CRD 1. Residues 35–37 form the intermolecular antiparallel β-sheet with gD (Carfi et al., 2001; Connolly et al., 2003). A systematic structure-based mutagenesis approach revealed that 17 residues in CRD 1 and 4 in CRD 2 are directly involved in the HVEM -gD interface. Some mutations completely abolish the HVEM binding to gD and its function as an HSV -1 receptor (Connolly et al., 2002).
Modified heparan sulphate
3-O-sulphated heparan sulphate represents the third identified HSV receptor, structurally unrelated to nectins and HVEM (Shukla et al., 1999). It serves as receptor for HSV -1, but not for unrestricted rid mutants or HSV -2. Thus, HSV -1 can use heparan sulphate GAGs not only for the initial step of attachment to the target cell, but it also recognizes some specifically modified sites on heparan sulphate (Shukla and Spear, 2001). The physical interaction with gD is in the range of 10−6 M, as measured by affinity co-electophoresis, and was not detectable by ELISA.
The sites on heparan sulphate recognized by gD are generated by heparan sulphate d-glucosaminyl 3-O-sulfotransferases (3-OSTs). 3-O-sulphates are rare substitutions in heparan sulphate, generated by at least six 3-OSTs isoforms identified in humans and mice, 3-OST-3A and 3-OST-3B, 3-OST-2, 3-OST-4 and 3-OST-5 (Shukla and Spear, 2001). Of note, 3-OST-3s are not receptors per se, consistent with the finding that gD does not bind to the purified enzymes; rather, they catalyze the substitutions, which, in turn, generate the receptors and render CHO cells susceptible to HSV.
The relevance of this potential receptor to HSV infection in human cell lines and in humans remains to be ascertained. In terms of distribution, the 3-OST-3s are broadly expressed isoforms in cells of different origins and tissues; 3-OST-2 and 3-OST-4 expression is mainly in the brain. 3-OST-5 expression is limited to the skeletal muscle.
Receptor preference and usage
The availability of multiple alternative receptors raises a number of questions, such as: What receptor is preferred in cells that coexpress both molecules? Do both serve as bona fide receptors in humans? Are they differentially distributed, or employed in different tissues? Limited information is available on these topics.
Two parameters that may guide receptor preference in cell cultures are the affinity of the binding and the cell surface density of the receptors. Neither appears to be relevant in the case of gD, since gD affinity to nectin1 and to HVEM is of the same order of magnitude (Krummenacher et al., 1998). Moreover, cells that appear to be nectin1-negative by fluorescent antibody cell sorting (e.g., EA -1 cells) can be infected and entry is inhibited by antibody to nectin1 (our unpublished result), implying that low density receptors suffice to mediate entry. The same phenomenon is observed with HVEM (Krummenacher et al., 2004).
As far as infection of animal and human tissues is concerned, nectin1 is expressed in nearly every neuron of adult mice, in rat sensory neurons, in some synapses, and serves as the primary receptor for HSV -1 infection in sensory neurons (Haarr et al., 2001; Mata et al., 2001; Richart et al., 2003). Nectin1 is also expressed in the epithelium of the human and murine vagina where it mediates HSV -1 and -2 entry in the genital mucosa of female hosts (Linehan et al., 2004). Nectin1-α is detected at the cell–cell adherens junctions in human skin (Matsushima et al., 2003). Cumulatively, these data are compatible with a major role of nectin1 in the infection of sensory neurons and mucoepithelia in vivo. The tissue distribution of HVEM suggests that it serves as the principal receptor for HSV in activated T lymphocytes, or in lymphoid organs (spleen and thymus), in liver and lung. However, these organs are not target of HSV in the natural history of infection, except in the rare cases of disseminated infection. Recent data show that several clinical/primary isolates can use both receptors, as laboratory strains do (Krummenacher et al., 2004). The fact that viruses of different origin retain the ability to use both receptors suggests that this is a requirement for successful infection and spread in the host (Krummenacher et al., 2004). The interaction with HVEM in particular may have a role in immune evasion (La et al., 2002).
For other viruses, formal proof that a given receptor plays a critical role in humans has rested on correlations between genetic defects in the locus encoding the receptor and a diminished susceptibility to viral infection. A rare truncation of the nectin1 gene has been identified. The phenotypes associated with loss of both alleles include cleft lip/palate, hidrotic ectodermal dysplasia (CLPED1), hair abnormalities, developmental defects of the hands and, in some cases, mental retardation. Studies on the serostatus of these patients for evidence of HSV infection have not been published. A few studies investigated nectin1, nectin2 and HVEM gene polymorphisms and correlations with HSV infection. At present, no relatively common polymorphism has been found to correlate with HSV serostatus or symptoms (Patera et al., 2002; Struyf et al., 2002; Krummenacher et al., 2004). Wild-type HSV can infect CLPED 1 fibroblasts through HVEM in vitro, indicating that the ability to exploit redundant receptors may be favorable to the virus in vivo as well (Krummenacher et al., 2004).
The animal orthologues of nectin and HVEM
HSV infects numerous mammalian species, some of which – mice, rabbits and primates – are extensively employed as animal models. The question arises whether they are faithful models in terms of receptor usage.
Porcine and bovine alphaherpesviruses promiscuously use human nectin1, implying that animal orthologs of nectins serve as receptors of these viruses in their hosts (Cocchi et al., 1998b; Geraghty et al., 1998; Warner et al., 1998). Indeed, nectins are conserved among mammals, and nectin1 orthologs have been found in cells derived from mice, hamsters, pigs, cows and monkeys, and nectin2 has a mouse ortholog, suggesting a conserved function in the evolution of these proteins (Milne et al., 2001). A HVEM ortholog is expressed in mice (Yoon et al., 2003). In turn, the murine orthologs of nectin1, nectin2, HVEM, and the porcine and bovine hortologs of human nectin1 can serve as species non-specific HSV receptors when transfected into receptor-negative cells (Menotti et al., 2000; Shukla et al., 2000; Menotti et al., 2001; Milne et al., 2001; Menotti et al., 2002a). The affinity of these potential receptors for gD ranges from as high as that of human nectin1, to very low or undetectable. Because the animal orthologs of nectin, and perhaps also of HVEM serve as receptors of HSV in mice and other animal species, these systems appear to relatively faithfully model HSV receptor usage in humans.
Site of HSV entry into the cell
It has been a long held paradigm that HSV enters cells by fusion at the plasma membrane. Recent evidence indicates that in some cells entry is through an endocytic pathway, and that both the cell type, and the structural features of the receptors are determinants that control the site of entry. Specifically, in cells like HeLa and CHO expressing nectin1 or HVEM, entry is inhibited by drugs that modify the pH of the endosomal compartment (low pH-sensitive entry) (Nicola et al., 2003). However, when nectin1 or HVEM are expressed in J cells, they mediate entry at the plasma membrane. Furthermore, when nectin1 is retargeted to endosomes, by means of a chimeric nectin1-EGFR (epidermal growth factor receptor) chimera, or is sorted to lipid rafts, by means of a nectin1-glycosylphosphatidylinositol anchor chimera, the pathway of entry into J cells becomes endocytic (Gianni et al., 2004). Of note, when HSV infects cells carrying wt-nectin1, neither nectin nor gD localize at lipid rafts, but gB does (Bender et al., 2003). All in all, HSV fusion glycoproteins are well suited to perform two quite different pathways of entry.
Role of gD-receptor interaction in triggering fusion
Crucial to our understanding of how HSV enters cells is the comprehension of how gD binding to its receptor triggers fusion. A hint has come from the unexpected observation that the soluble gD ectodomain is both necessary and sufficient to rescue the infectivity of the non infectious gDnull HSV mutant (Cocchi et al., 2004). Entry mediated by soluble gD requires not only the N-terminus, carrying the receptor-binding sites, but also the C-terminus carrying the pro-fusion domain, required to trigger fusion but not for receptor binding. These findings, together with the observation that a glycosylphosphatidylinositol-anchored form of gD, or substitution of the transmembrane and cytoplasmic tail with heterologous regions leave gD function unaltered (Browne et al., 2003) demonstrate that the transmembrane region and cytoplasmic tail do not play any demonstrable function, except to ensure that gD is delivered to the gD receptor along with the virion, and argue that the role of gD in HSV entry is to signal receptor-recognition to the downstream glycoproteins and to trigger fusion (Cocchi et al., 2004).
Biochemical and structural studies indicate that the receptor-mediated activation of gD takes the form of a conformational change (Cocchi et al., 2004; Fusco et al., 2005; Krummenacher et al., 2005). In the unliganded state the virion gD adopts a conformation in which the flexible C terminus folds back, wraps the N-terminus and masks the receptor binding sites. At receptor binding, the C-terminus is displaced from its binding site on the N-terminus, the receptor binding sites are unmasked and become occupied by the receptor. The binary complex made of receptor plus gD with the displaced C-terminus must create a surface suitable for gB and gH-gL recruitment.
Execution of membrane fusion and its control
gB, gH, gL are essential for entry of all herpesviruses, since they are conserved among all human herpesviruses, with the highest extent of sequence conservation seen in gB. Heterodimer formation between gH and gL is also a conserved feature amongst herpesviruses. Altogether, gB, gH and gL appear to be the executors of fusion and constitute the conserved fusion machinery across the herpesvirus family.
Critical properties of gH and gB have been elucidated recently, and provide an intriguing scenario. On one hand, molecular and biochemical analyses of gH highlighted properties typical of class 1 fusion glycoproteins. Because the gH structure has not yet been solved, these properties wait for confirmation at the structural level. On the other hand, the crystal structure of gB has been solved, it exhibits a remarkable similarity to that of vesicular stomatitis G protein, and to viral fusion glycoproteins in general. Biochemical and mutational confirmation are still to be provided. At present, a most likely scenario is that both gB and gH⋅gL are fusion executors. How the two glycoproteins cooperate to execute fusion, and why two, and not one, fusion executors are required in the herpesviridae family is unclear. It is worthwhile to note that entry by fusion at plasma membrane, and entry by fusion in endocytic vesicles require all four glycoproteins (gD, gB, gH and gL) (Nicola et al., 2003; Nicola and Straus, 2004). These requirements rule out the possibility that gB serves as fusion executor in one cellular compartment, and gH⋅gL serves as fusion executor in another cell compartment.
The glycoproteins that execute fusion
gH –gL
gH is a type-1 virion glycoprotein encoded by the UL 22 gene (Gompels and Minson, 1986). Soon after its discovery, it was recognized as an essential glycoprotein for virion infectivity, as its deletion produced non infectious progeny and abolished cell–cell fusion (Forrester et al., 1992). Neutralizing antibodies to gH block virus entry but permit attachment, indicating a role at a post-attachment step (Fuller et al., 1989). gH appears to contain elements associated with fusion of membranes, i.e. a hydrophobic α-helix 1 (residues 377-397) with properties typical of a fusion peptide and two heptad repeats with propensity to form a coiled coil. α-Helix 1 is positionally conserved in all the gH orthologs across the herpesviridae family; in HSV -2 it is located in a loop made of two cysteins. α-Helix 1 is able to interact with biological membranes, can convert a soluble glycoprotein (gD amino acid residues 1-260) into a membrane-bound glycoprotein, and can be functionally replaced by fusion peptides derived from glycoproteins of other, unrelated viruses (Gianni et al., 2005a). A peptide with the sequence of α-helix 1 induces fusion of liposomes and exhibits a strong flexibility documented as ability to adopt an α-helical conformation (Galdiero, S. et al., 2006; Gianni et al., 2006a). These properties strongly argue in favor of α-helix 1 as a candidate fusion peptide loop. Two heptad repeats, capable to form coiled coils and to interact with each other, form a structure of increased α-helical content and are potentially suitable to form a six-helix bundle (Gianni et al., 2005b; Galdiero, S. et al., 2006; Gianni et al., 2006b). Additional elements in gH are a second predicted α-helical domain of lower hydrophobicity than the candidate fusion peptide (aa 513-531), and a pre-transmembrane sequence (aa 626-644) with predicted propensity to partition at membrane interface (Galdiero, S. et al., 2006; Gianni et al., 2006a).
Synthetic peptides corresponding to the heptad repeats inhibit virus infection if present at the time of virus entry into the cell (Gianni et al., 2005b; Galdiero, S. et al., 2006; Gianni et al., 2006b). The presence of coiled coil motifs predicts that gH must undergo profound conformational changes at fusion. Because fusion peptides and coiled coil heptad repeats represent characteristic functional domains in type 1 viral fusion glycoproteins, gH is a candidate fusion executor in HSV.
It remains to be determined whether gH interacts with cellular receptors. The interaction with an integrin is not critical given that mutagenesis of a RGD motif did not reduce virus entry and cell fusion (Galdiero, M. et al., 1997). It is of interest that the transmembrane and C-terminal tail regions of gH can not be exchanged with those of heterologous proteins, in contrast with what happens with gD (Harman et al., 2002; Jones and Geraghty, 2004).
A ts mutant, tsQ26, exhibited a phenotype characterized by the production of non-infectious extracellular virions, along with the intracellular retention of gH and of infectious virions (Desai et al., 1988). This phenotype suggested a peculiar mechanism of intracellular retention of gH. A clue to understanding the intracellular trafficking of gH came from the observations that, when expressed from a transgene, gH had a Mr lower than that of mature gH, was not transported to the cell surface, and was retained in the ER unless the cells were superinfected (Gompels and Minson, 1986; Foà-Tomasi et al., 1991; Roberts et al., 1991). The gene product required for gH trafficking and maturation, identified by Johnson and coworkers, is gL (Hutchinson et al., 1992a); gL is required for proper folding and traffiking of gH in all human Herpesviruses (Kaye et al., 1992; Liu et al., 1993).
gL is a soluble glycoprotein encoded by UL 1 gene; its presence in the virion envelope is ensured by complex formation with gH (Hutchinson et al., 1992a). In accordance with gH attributes, an HSV mutant unable to express gL could not enter cells, and its particles lacked glycoprotein H (Roop et al., 1993). Both gH and gL are required for fusion in the cell–cell fusion assay (Turner et al., 1998). The first 323 amino acids of gH and the first 161 amino acids of gL can form a stable secreted hetero-oligomer, while the first 648 amino acids of gH are required for reactivity to conformation-dependent antibodies, indicative of correct conformation and oligomerization (Peng et al., 1998). gL is a locus of a syn mutation, confirming a role of the gH-gL hetero-oligomer in HSV fusion. The exact role of gL in fusion remains to be elucidated. Because its binding site on gH maps both upstream and downstream of the hydrophobic α-helix, it has been proposed that its role may be to shield the gH hydrophobic sequence, and thus to enable gH water solubility and solvent interface (Gianni et al., 2005a).
gB
gB plays two opposite roles in fusion, i.e. it participates in fusion execution, and it exerts anti-fusion activity. The two functions are physically separated and reside in the ectodomain and the cytoplasmic tail, respectively. gB is a type-1 virion glycoprotein encoded by the UL 27 gene (Bzik et al., 1984; Pellett et al., 1985). Its crystal structure reveals a trimer with a coiled coil core. Remarkably, its structure resembles closely that of vesicular stomatitis virus G protein (Heldwein et al., 2006; Roche et al., 2006). Despite the facts that a canonical fusion peptide has not been detected by biochemical, molecular or structural analyses, and that the region homologous to the fusion peptide loop in vesicular stomatitis virus G protein appears to be suboptimal for membrane insertion, the structural similarity between gB and vesicular stomatitis virus G protein strongly relates gB to viral fusion glycoproteins. It has been proposed that the two glycoproteins may represent a novel class of fusion glycoproteins (Heldwein et al., 2006; Roche et al., 2006).
From a structural point of view, gB is a trimeric spike. Each of the three protomers (residues 103–730) appears to be composed of five distinct domains (named Ⅰ-Ⅴ), displaying multiple contact sites (Heldwein et al., 2006). Domain Ⅰ, the “base”, is a continous chain with a fold typical of pleckstrin homology domains. Domain Ⅱ, the “middle”, is made of two discontinuous segments, forming a structure reminiscent of a pleckstrin homology superfold. Domain Ⅲ, the “core”, comprises three discontinuous segments: its prominent feature is a 44-residue α-helix that forms the central coiled coil with its trimeric counterparts. Domain Ⅳ, the “crown”, adopts a novel structure, and is fully exposed on top of the trimeric spike. Domain Ⅴ, the “arm”, is a long extension spanning the full length of the protomer. Of note, its residues do not contact residues of the same protomer, but instead accommodate into the groove formed by the “cores” of the other two protomers.
The role of gB in virion infectivity and cell–cell fusion is inferred by numerous lines of evidence, including (ⅰ) the phenotype of a gB deletion mutant virus which produces non-infectious particles, (ⅱ) the neutralizing activity of antibodies to gB, (ⅲ) gB as a genetic locus of syncytial mutations, and (ⅳ) the requirement for gB in the cell-cell fusion assay (Manservigi et al., 1977; Cai, W. Z. et al., 1987; Turner et al., 1998). Functional domains in the ectodomain were identified by means of two sets of mutations: temperature sensitive mutations for viral growth, and resistance to antibodies with potent neutralizing activity. The first ones, exemplified by the mutants tsB5 and tsJ12, reside in the gB ectodomain, confer a temperature-sensitive phenotype, and affect the rate of virus entry (Bzik et al., 1984). Likely, these mutations affect the gB domain involved in execution of fusion. Following the determination of gB crystal structure, it was recognized that the epitopes of potent neutralizing antibodies, either centered around single amino acid residues or formed by continuous regions, reside on the trimer surface, on the lateral faces of the spike or on the tip of the crown (Pellett et al., 1985; Kousoulas et al., 1988; Highlander et al., 1989; Pereira et al., 1989; Qadri et al., 1991; Heldwein et al., 2006).
The quartet of gD, gB, gH and gL assemble into a complex at virus entry
The nature of the interactions between the complex formed by gD plus its receptor and the executors of fusion is critical to understand the mechanisms by which HSV (and by extension all other herpesviruses) enter cells.
The quartet of glycoproteins essential for HSV entry and fusion (gD, gB, gH and gL) assemble into a complex at virus entry and in infected cells. Complex assembly strictly requires one of the gD receptors, either nectin1 or HVEM. The same complex is assembled also in cells transfected with the quartet, implying that no additional viral protein other than those that participate in the complex itself is required. Because the complex is assembled at virus entry and in transfected cells committed to form polykaryocytes, and fails to be assembled in the absence of either a receptor to gD or of gD, complex assembly appears to be a critical step in the process of virus entry and fusion.
The proteins that negatively control fusion
Cells infected with wt-virus do not form syncytia, despite the fact that they express the fusion glycoproteins at their surface. Syncytia are only formed when the virus carries one of the syncytial (syn) mutations, which map to genes encoding gB, gL, gK, UL 24, or UL 20. By contrast, cells expressing the quartet of gB, gD, gH and gL readily form syncytia (Turner et al., 1998). The paradox may be explained by assuming that the wt-alleles of proteins that are target of syn mutations exert a negative control on fusion. This has, in fact, been verified for gB, gK, and UL 20 (Fan et al., 2002; Avitabile et al., 2003; Avitabile et al., 2004). As outlined below, HSV has evolved at least two mechanisms by which it blocks fusion. One is exerted through downmodulation of gB cell surface expression, the other is exerted through the concerted action of gK and UL 20p. Still other proteins (UL24 and UL 45) are likely to exert anti-fusion activity. The evolution of functional redundancy implies that uncontrolled fusion is inimical to HSV-1 replication and spread in nature, and therefore the virus needs to exert a tight control on it.
gB
The anti-fusion activity of gB is located in the cytoplasmic tail, which carries at least two physically distinct functional domains: the syn mutation and the endocytosis motifs. Each of them, separately, appears to reduce fusion. Structurally, the cytoplasmic tail carries two predicted α-helices. The syn mutations are located immediately downstream of the most N-terminal α-helix. Embedded in the region of the C-terminal α-helix is one, and possibly two functional endocytosis motifs (YTQV889–892 and LL 871 (Fan et al., 2002; Avitabile et al., 2004; Beitia Ortiz de Zarate et al., 2004). Deletion of the membrane-proximal α-helix abrogates virus infectivity, implying that this region is critical. Its role, and the molecular mechanism of the syn3 mutation remain to be elucidated. The membrane-distal α-helix is also implicated in the negative control of fusion, since its deletion increases fusion in the cell fusion assay, and confers a syncytial phenotype upon virus-infected cells (Foster, et al., 2001a; Avitabile et al., 2004). Its antifusion activity is mainly exerted through endocytosis, which acts to decrease the steady state amounts of gB from the cell surface, such that gB becomes a limiting factor in fusion. Of note, the gB-decorated endocytosis vesicles-vacuoles represent the hallmark of gB localization in infected cells.
gK
gK is a polytopic glycoprotein encoded by the UL 53 gene, whose topology is still debated (Hutchinson et al., 1992b; Foster, et al., 2001b). It carries an N-terminal extracellular domain, and two or three TM regions connected by loops (Foster, et al., 2003b). Its hydrophobicity, poor immunogenicity and overall problems in its detection have made this glycoprotein a difficult one to study. In infected cells, gK localizes mainly to the Golgi apparatus. One controversial aspect is whether it localizes to the plasma membranes and to virions. When expressed from a transgene, gK is primarily located is at the ER (Hutchinson et al., 1992b; Avitabile et al., 2003; Foster, et al., 2003a). When coexpressed with UL 20, both proteins localize to the Golgi apparatus (Avitabile et al., 2004).
gK exerts anti-fusion activity in the cell–cell fusion assay (Avitabile et al., 2003). Mutant viruses carrying a partial or a complete deletion in the gK gene have two major phenotypes (Hutchinson and Johnson, 1995; Foster, and Kousoulas, 1999). First, they form syncytia, arguing that the anti-fusion activity is exerted also in the context of infected cells. Second, they are defective in virus egress, arguing that the anti-fusion activity of gK is exerted not only at the plasma membrane, but also in the membranes of the exocytic compartment. This would provide an explanation as to why these membranes are heavily decorated with fusion glycoproteins, yet do not fuse one with the other. According to this model, the gK role in virion egress may be exerted by maintaining a functional exocytic pathway. It should be stressed that, if indeed gK is also a virion constituent, then, at virus entry into the cell, the trigger to fusion must simultaneously relieve the block to fusion exerted by gK (Avitabile et al., 2004).
UL20
UL20p is a polytopic unglycosylated protein with several analogies to gK. Its hydrophobicity and scarce immunogenicity have hampered its characterization. UL 20p is predicted to carry 4 transmembrane segments (McGeoch et al., 1988; Melancon et al., 2004). In the infected cells UL 20p localizes at the Golgi apparatus and the nuclear membranes, and is not detectable at the plasma membrane. It has not been detected in virions. When expressed from a transgene, UL 20p predominant localization is at the ER (Avitabile et al., 1994; Ward et al., 1994). It relocalizes to the Golgi apparatus, when coexpressed with gK (Foster, et al., 2003b; Avitabile et al., 2004).
Two mutant viruses deleted in UL 20 gene have been constructed, both of which are highly defective in secretion of virions to the extracellular space (Baines et al., 1991; Foster, et al., 2004). The first deletion virus was subsequently reported to carry an in-frame fusion between UL 20.5 (not known at the time the deletion virus was constructed) and the C-terminus of UL 20 gene, and was characterized by syncytia formation and by the entrapping of virions in the perinuclear space (i.e., the space between the inner and outer nuclear membranes) – a phenotype particularly evident in cells whose Golgi apparatus became fragmented following infection (Baines et al., 1991). This phenotype can be interpreted as indication that the UL 20p exerts a negative control on fusion. Cells infected with the second deletion virus showed enveloped virions as well as unenveloped nucleocapsids accumulating in the cytoplasm, and occasionally virion envelopes containing multiple capsids within intracytoplasmic vacuoles. These phenotypes were also interpreted to mean that UL 20p acts as an inhibitor of membrane fusion, and, interestingly, that UL 20p may act to maintain a single nucleocapsid for each envelope and to prevent fusion of enveloped virions among themselves (Foster, et al., 2004). The complexity of these phenotypes reflects both direct and indirect effects of UL 20p, including the role of UL 20p in the intracellular transport of gK and possibly of the fusion glycoproteins.
The possibility that UL 20p exerts an anti-fusion activity was probed in the cell–cell fusion assay, which showed a block to fusion in cells coexpressing UL 20p and gK, but not in cells expressing UL 20 alone. The block was cell line dependent (Avitabile et al., 2004). The similar behavior of gK and UL 20p, their colocalization, their mutual ability to influence each other localization, and their concerted anti-fusion activity make it likely that the two proteins act in a complex, and that they share a common target.
Nucleocapsid transport to the nuclear pore
Virus entry culminates in the release of capsids and approximately twenty tegument proteins into the cytosol. The capsids and some of the tegument proteins, e.g., αTIF, travel to the nuclear pore. Since diffusion of molecules larger than 500 kDa is restricted in the cytoplasm, viruses and nucleocapsids require a transport system. This is particularly true for neurotropic viruses that travel long distances in the axon during retrograde or anterograde transport (Enquist et al., 1998). It has been calculated that in the absence of an active transport mechanism, it would take a herpes virus capsid 231 years to diffuse 10 mm in the axonal cytoplasm (Sodeik, 2000).
Microtubules represent the cytoplasmic highways on which HSV is transported (reviewed in Döhner and Sodeik, 2005). At virus entry, capsids co-localize with microtubules, and their depolymerization reduces capsid transport to the nucleus (Sodeik et al., 1997; Mabit et al., 2002). Microtubules are polar structures with fast growing plus-ends typically localized in the cell periphery, and less dynamic minus-ends that are usually anchored in close proximity of the nucleus at the microtubule organizing centre (MTOC). Molecular motors use ATP -driven conformational changes to transport cargo along microtubules. Transport to the plus-ends is catalyzed by kinesins and that to minus-end by cytoplasmic dynein and dynactin (Döhner and Sodeik, 2005). Dynein and dynactin mediate capsid transport to the cell centre, since incoming capsids colocalize with these motors (Sodeik et al., 1997; Döhner et al., 2002), and overexpression of dynamitin, a subunit of the dynactin complex, inhibits capsid transport (Döhner et al., 2002). How capsids move further from MTOC to the nuclear pore complex is unclear.
Analysis of HSV -1 entry by digital time-lapse fluorescence microscopy showed that GFP -tagged capsids can move along microtubules both towards and away from the nucleus, with maximal speeds of 1.1 µm/s. The transport is saltatory and bidirectional, but in neuronal processes it shows a retrograde bias towards the cell body (Smith et al., 2001). Efforts are underway to identify the virion proteins that may interact with kinesins as well as dynein or dynactin. Two candidates are UL 34p, which, however, is absent from mature virions, and US 11, which appears to bind the heavy chain of conventional kinesin (Diefenbach et al., 2002). Once the capsids have reached the proximity of the nucleus, they seem to bind to filaments emanating from the nuclear pores (Batterson et al., 1983; Sodeik et al., 1997). This docking is believed to induce capsid destabilization, release of the viral DNA, and its translocation through the nuclear pore into the nucleoplasm (Ojala et al., 2000). Temperature-sensitive mutants in the UL 36 gene accumulate filled viral capsids at the nuclear pore complexes at the non-permissive temperature, suggesting that the large tegument protein VP 1–3 is involved in uncoating of the viral genome (Batterson et al., 1983; Ojala et al., 2000).
VZV
There are several remarkable differences between VZV and HSV entry. Because the respective viral glycoproteins undoubtedly influence these differences, the VZV gene products will be briefly summarized.
Is VZV gE a substitute for functions of HSV gD?
All but one of the proteins that have been illustrated above for HSV have a counterpart in VZV. For those glycoproteins for which sufficient information is available, a substantial functional similarity is observed. The single most notable difference between VZV and HSV in terms of the glycoproteins is the absence of gD in the VZV genome. At the same time, VZV is well suited for cell-to-cell spread, which takes place by fusion of the infected cell with an adjacent uninfected cell, whereas HSV is better suited for virion-to-cell infection (at least in cultured cells). So, it is tempting to speculate that the absence of a VZV gD gene may contribute to these differences.
Despite the fact that gD plays such a pivotal role in HSV entry, gD is not conserved throughout the alphaherpesviruses. In the porcine herpesvirus PrV, gD is required for virion-to cell infectivity but not for cell-to-cell spread of the virus. Consistently, gD is not a requirement for the PrV cell-cell fusion assay, although its presence greatly enhances fusion efficiency. Assuming that common basic mechanisms are shared by all of the alphaherpesviruses and given that the triplet gH-gL-gB is conserved, the question then arises: which VZV glycoprotein substitutes for the functions encoded in HSV gD, i.e., receptor recognition and triggering of fusion. The two functions might well be distributed over different entities, but a trigger to fusion consequent to virion interaction with a receptor appears to be essential.
In VZV, four glycoproteins are known to be essential. They are gB, gH, gL and gE (Keller et al., 1984; Montalvo and Grose, 1986; Forghani et al., 1994; Duus et al., 1995; Mallory et al., 1997; Mo et al., 2002). Of note, the HSV gE gene lies in the S component of the genome, proximal to gD; as stated above, VZV lacks the gD gene. Instrumental to our understanding of the role of gE are the results of VZV glycoprotein cell–cell fusion assays. In transfected cells, fusion is induced by coexpression of either gH-gL or of gB-gE (Duus et al., 1995; Duus and Grose, 1996; Maresova et al., 2001). With regard to genome stability, VZV is considered to be one of the more genetically stable herpesviruses. However, viral mutants carrying missense mutations in the gE ectodomain are being isolated from humans; one of them is more fusogenic in cell cultures and in the SCID -hu mice (Santos et al., 1998, 2000). Cumulatively, both circumstantial and genetic evidence supports the possibility that VZV gE subsumes at least some of roles of HSV gD.
Endocytosis of the VZV glycoproteins gE, gB, gH, and the negative regulation of fusion
Three VZV glycoproteins carry functional tyrosine-based endocytosis motifs; they are gE, gB and gH.
The gE cytoplasmic tail has a YAGL sequence beginning with a tyrosine residue 582. As determined by mutagenesis studies, the tyrosine residue is part of a conserved YXXL endocytosis motif. The internalized gE trafficks to the trans-Golgi or is recycled to the cell surface. In addition, the C-tail also contains phosphorylation sites (Kenyon et al., 2002). It has been suggested that serine/threonine and tyrosine phosphorylation of gE may serve as sorting signals for internalized receptors and that formation of a gE–gI complex facilitates gE endocytosis (Olson and Grose, 1997; Olson et al., 1998).
VZV gB contains three predicted endocytosis motifs within its cytoplasmic domain: YMTL (aa 818–821), YSRV (aa 857–860), and LL (aa 841–842). Both tyrosine-based motifs mediate gB internalization, but only the YSRV motif is absolutely required for endocytosis. The YMTL motif functions in trafficking of internalized gB to its subsequent localization in the trans Golgi. The third potential endocytosis motif is a dileucine sequence, whose function is under study (Heineman and Hall, 2001). Of note, VZV gI, the partner of VZV gE, also contains a dileucine endocytosis motif in its C-tail (Olson and Grose, 1998).
Like VZV gE and gB, VZV gH contains a functional but previously unrecognized tyrosine based YNKI motif in its short cytoplasmic tail, which mediates clathrin-dependent and antibody-independent endocytosis. Alignment analysis of the VZV gH cytoplasmic tail with other herpesvirus gH homologues reveals two interesting features: (ⅰ) herpes simplex virus types 1 and 2 homologues lack an endocytosis motif while all other alphaherpesvirus gH homologues contain a potential motif, and (ⅱ) the VZV gH C-tail is actually longer than predicted in the original sequence analysis and thus can provide the proper context for a functional endocytosis motif (Pasieka et al., 2003). Surprisingly, the endocytosis-deficient VZV gH mutant plasmid effects greater cell-cell fusion than the wild-type gH plasmid. This result leads to the conclusion that VZV gH endocytosis represents a mechanism through which cell–cell fusion is negatively regulated, i.e., by modulating the amount of fusogenic gH on the cell surface (Pasieka et al., 2004). In this respect, therefore, VZV gH shares a basic mechanism of negative regulation of fusion with HSV gB.
Cumulatively, this comparison of the VZV and HSV -1 systems is very instructive as it highlights that both viruses have evolved an essentially similar mechanism of control of fusion, based on endocytosis and consequent limitation of cell surface expression of the fusion executors themselves. A notable difference between the two viruses is that this type of control appears to be is exerted in HSV -1 mainly by gB and in VZV mainly by gH.
Acknowledgments
We are grateful to Drs. Roselyn Eisenberg, Joel Baines, Charles Grose, and Beate Sodeik for critical reading of this chapter.
References
- Avitabile E., Ward P. L., Lazzaro, Torrisi M. R., Roizman B., Fiume G., and Campadelli-1994The herpes simplex virus UL20 protein compensates for the differential disruption of exocytosis of virions and membrane glycoproteins associated with fragmentation of the Golgi apparatus J. Virol. 687397–7405. [PMC free article: PMC237182] [PubMed: 7933123]
- Avitabile E., Lombardi G., Fiume G., and Campadelli-2003Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell–cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH and gL J. Virol. 776836–6844. [PMC free article: PMC156197] [PubMed: 12768003]
- Avitabile E., Lombardi G., Gianni T., Capri M., Fiume G., and Campadelli-2004Coexpression of UL20p and gK inhibits cell–cell fusion mediated by herpes simplex virus glycoproteins gD, gH-gL, and wt-gB or an endocytosis-defective gB mutant, and downmodulates their cell surface expression J. Virol. 788015–8025. [PMC free article: PMC446093] [PubMed: 15254173]
- Baines J. D., Ward P. L., Fiume G., Roizman B., Campadelli-1991The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress J. Virol. 656414–6424. [PMC free article: PMC250678] [PubMed: 1719228]
- Batterson W., Furlong D., Roizman B. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 1983;45:397–407. [PMC free article: PMC256421] [PubMed: 6296445]
- Beitia Ortiz de Zarate I., Kaelin K., Rozenberg F. Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J. Virol. 2004;78:1540–1551. [PMC free article: PMC321396] [PubMed: 14722308]
- Bender F. C., Whitbeck J. C., Ponce de Leon M., Lou H., Eisenberg R. J., Cohen G. H. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 2003;77:9542–9552. [PMC free article: PMC187402] [PubMed: 12915568]
- Browne H., Bruun B., Whiteley A., Minson T. Analysis of the role of the membrane-spanning and cytoplasmic tail domains of herpes simplex virus type 1 glycoprotein D in membrane fusion. J. Gen. Virol. 2003;84:1085–1089. [PubMed: 12692272]
- Bzik D. J., Fox B. A., DeLuca N. A., Person S. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology. 1984;137:185–190. [PubMed: 6089415]
- Cai W. Z., Person S., Warner S. C., Zhou J. H., DeLuca N. A. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 1987;61:714–721. [PMC free article: PMC254011] [PubMed: 3027398]
- Cai W. Z., Gu B., Person S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion [published erratum appears in J. Virol. 1988 Nov; 62(11):4438] J. Virol. 1988;62:2596–2604. [PMC free article: PMC253689] [PubMed: 2839688]
- Fiume G., Arsenakis M., Farabegoli F., Roizman B.Campadelli-1988Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus J. Virol. 62159–167. [PMC free article: PMC250514] [PubMed: 2824844]
- Fiume G., Stirpe D., Boscaro A., et al. Campadelli-1990Glycoprotein C-dependent attachment of herpes simplex virus to susceptible cells leading to productive infection Virology 178213–222. [PubMed: 2167550]
- Fiume G., Cocchi F., Menotti L., Lopez M.Campadelli-2000The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells Rev. Med. Virol. 10305–319. [PubMed: 11015742]
- Carfi A., Willis S. H., Whitbeck J. C., et al. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell. 2001;8:169–179. [PubMed: 11511370]
- Cocchi F., Lopez M., Menotti L., Aoubala M., Dubreuil P., Fiume G., and Campadelli-1998aThe V domain of herpesvirus Ig-like receptor (HIgR) contains a major functional region in herpes simplex virus-1 entry into cells and interacts physically with the viral glycoprotein D Proc. Natl Acad. Sci. USA 9515700–15705. [PMC free article: PMC28107] [PubMed: 9861033]
- Cocchi F., Menotti L., Mirandola P., Lopez M., Fiume G., and Campadelli-1998bThe ectodomain of a novel member of the immunoglobulin superfamily related to the poliovirus receptor has the attibutes of a bonafide receptor for herpes simplex viruses 1 and 2 in human cells J. Virol. 729992–10002. [PMC free article: PMC110516] [PubMed: 9811737]
- Cocchi F., Lopez M., Dubreuil P., Fiume G., Menotti L., Campadelli-2001Chimeric nectin1-poliovirus receptor molecules identify a nectin1 region functional in herpes simplex virus entry J. Virol. 757987–7994. [PMC free article: PMC115042] [PubMed: 11483743]
- Cocchi F., Fusco D., Menotti L., et al. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl Acad. Sci. USA. 2004;101:7445–7450. [PMC free article: PMC409938] [PubMed: 15123804]
- Connolly S. A., Whitbeck J. J., Rux A. H., et al. Glycoprotein D homologs in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human HveC(nectin-1) with different affinities. Virology. 2001;280:7–18. [PubMed: 11162814]
- Connolly S. A., Landsburg D. J., Carfi A., Wiley D. C., Eisenberg R. J., Cohen G. H. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol. 2002;76:10894–10904. [PMC free article: PMC136654] [PubMed: 12368332]
- Connolly S. A., Landsburg D. J., Carfi A., Wiley D. C., Cohen G. H., Eisenberg R. J. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/ HVEM binding interface. J. Virol. 2003;77:8127–8140. [PMC free article: PMC161942] [PubMed: 12829851]
- Connolly S. A., Landsburg D. J., Carfi A., et al. Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J. Virol. 2005;79:1282–1295. [PMC free article: PMC538551] [PubMed: 15613355]
- Davison A. J., Scott J. E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986;67(9):1759–1816. [PubMed: 3018124]
- Desai P. J., Schaffer P. A., Minson A. C. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J. Gen. Virol. 1988;69(6):1147–1156. [PubMed: 2838568]
- Diefenbach R. J., Saksena M., Diefenbach E., et al. , Miranda-2002Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain J. Virol. 763282–3291. [PMC free article: PMC136023] [PubMed: 11884553]
- hner K., Sodeik B.Dö2005The role of the cytoskeleton during viral infection Curr. Top. Microbiol. Immunol. 28567–108. [PubMed: 15609501]
- hner K., Wolfstein A., Prank U., et al. Dö2002Function of dynein and dynactin in herpes simplex virus capsid transport Mol. Biol. Cell 132795–2809. [PMC free article: PMC117943] [PubMed: 12181347]
- Dolter K. E., Goins W. F., Levine M., Glorioso J. C. Genetic analysis of type-specific antigenic determinants of herpes simplex virus glycoprotein C. J. Virol. 1992;66:4864–4873. [PMC free article: PMC241321] [PubMed: 1378512]
- Duus K. M., Grose C. Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J. Virol. 1996;70:8961–8971. [PMC free article: PMC190993] [PubMed: 8971025]
- Duus K. M., Hatfield C., Grose C. Cell surface expression and fusion by the varicella-zoster virus gH:gL glycoprotein complex: analysis by laser scanning confocal microscopy. Virology. 1995;210:429–440. [PubMed: 7618278]
- Enquist L. W., Husak P. J., Banfield B. W., Smith G. A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998;51:237–247. [PubMed: 9891589]
- Fan Z., Grantham M. L., Smith M. S., Anderson E. S., Cardelli J. A., Muggeridge M. I. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell–cell fusion, but these properties are unrelated. J. Virol. 2002;76:9271–9283. [PMC free article: PMC136473] [PubMed: 12186911]
- Tomasi L., Avitabile E., Boscaro A., et al. Foà-1991Herpes simplex virus (HSV) glycoprotein H is partially processed in a cell line that expresses the glycoprotein and fully processed in cells infected with deletion or ts mutants in the known HSV glycoproteins Virology 180474–482. [PubMed: 1846486]
- Forghani B., Ni L., Grose C. Neutralization epitope of the varicella-zoster virus gH:gL glycoprotein complex. Virology. 1994;199:458–462. [PubMed: 7510086]
- Forrester A., Farrell H., Wilkinson G., Kaye J., Poynter, Minson T. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 1992;66:341–348. [PMC free article: PMC238293] [PubMed: 1309250]
- Foster T. P., Kousoulas K. G. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 1999;73:8457–8468. [PMC free article: PMC112865] [PubMed: 10482598]
- Foster T. P., Melancon J., Kousoulas K. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology. 2001a;287:18–29. [PubMed: 11504538]
- Foster T. P., Rybachuk G. V., Kousoulas K. G. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 2001b;75:12431–12438. [PMC free article: PMC116139] [PubMed: 11711633]
- Foster T. P., Alvarez X., Kousoulas K. G. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 2003a;77:499–510. [PMC free article: PMC140622] [PubMed: 12477855]
- Foster T. P., Rybachuk G. V., Alvarez X., Borkhsenious O., Kousoulas K. G. Overexpression of gK in gK-transformed cells collapses the Golgi apparatus into the endoplasmic reticulum inhibiting virion egress, glycoprotein transport, and virus-induced cell fusion. Virology. 2003b;317:237–252. [PubMed: 14698663]
- Foster T. P., Melancon J. M., Baines J. D., Kousoulas K. G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004;78:5347–5357. [PMC free article: PMC400383] [PubMed: 15113914]
- Fuller A. O., Santos R. E., Spear P. G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 1989;63:3435–3443. [PMC free article: PMC250919] [PubMed: 2545914]
- Fusco D., Forghieri C., Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl Acad. Sci. USA. 2005;102:9323–9328. [PMC free article: PMC1166633] [PubMed: 15972328]
- Galdiero M., Whiteley A., Bruun B., Bell S., Minson T., Browne H. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol. 1997;71:2163–2170. [PMC free article: PMC191323] [PubMed: 9032350]
- Galdiero S., Vitiello M., D’Isanto M., et al. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 2006;87:1085–1097. [PubMed: 16603508]
- Geraghty R. J., Krummenacher C., Cohen G. H., Eisenberg R. J., Spear P. G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. [PubMed: 9616127]
- Gerber S. I., Belval B. J., Herold B. C. Differences in the role of glycoprotein C of HSV-1 and HSV-2 in viral binding may contribute to serotype differences in cell tropism. Virology. 1995;214:29–39. [PubMed: 8525631]
- Gianni T., Fiume G., Menotti L., Campadelli-2004Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes J. Virol. 7812268–12276. [PMC free article: PMC525084] [PubMed: 15507614]
- Gianni T., Martelli P. L., Casadio R., Fiume G., and Campadelli-2005aThe ectodomain of herpes simpex virus glycoprotein H contains a membrane alpha-helix with attributes of an internal fusion peptide, positionally conserved in the Herpesviridae family J. Virol. 792931–2940. [PMC free article: PMC548475] [PubMed: 15709012]
- Gianni T., Menotti L., Fiume G., and Campadelli-2005bA heptad repeat in herpes simplex virus gH, located downstream of the alpha-helix with attributes of a fusion peptide, is critical for virus entry and fusion J. Virol. 797042–7049. [PMC free article: PMC1112143] [PubMed: 15890943]
- Gianni T., Fato R., Bergamini C., Lenaz G., Campadelli-Fiume G. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol. 2006a;80:8190–8198. [PMC free article: PMC1563806] [PubMed: 16873275]
- Gianni T., Piccoli A., Bertucci C., Campadelli-Fiume G. Heptad repeat 2 in herpes simplex virus-1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 2006b;80:2216–2224. [PMC free article: PMC1395405] [PubMed: 16474129]
- Gompels U., Minson A. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology. 1986;153:230–247. [PubMed: 3016991]
- Griffiths A., Renfrey S., Minson T. Glycoprotein C-deficient mutants of two strains of herpes simplex virus type 1 exhibit unaltered adsorption characteristics on polarized or non-polarized cells. J. Gen. Virol. 1998;79(4):807–812. [PubMed: 9568976]
- Gruenheid S., Gatzke L., Meadows H., Tufaro F. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J. Virol. 1993;67:93–100. [PMC free article: PMC237341] [PubMed: 8380101]
- Grunewald K., Desai P., Winkler D. C., et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 2003;302:1396–1398. [PubMed: 14631040]
- Haarr L., Shukla D., Rodahl E., Canto, Spear P. G. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology. 2001;287:301–309. [PubMed: 11531408]
- Harman A., Browne H., Minson T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 2002;76:10708–10716. [PMC free article: PMC136627] [PubMed: 12368313]
- Harrop J. A., Reddy M., Dede K., et al. Antibodies to TR2 (herpesvirus entry mediator), a new member of the TNF receptor superfamily, block T cell proliferation, expression of activation markers, and production of cytokines. J. Immunol. 1998;161:1786–1794. [PubMed: 9712045]
- Heineman T. C., Hall S. L. VZV gB endocytosis and Golgi localization are mediated by YXXphi motifs in its cytoplasmic domain. Virology. 2001;285:42–49. [PubMed: 11414804]
- Heldwein E. E., Lou H., Bender F. C., Cohen G. H., Eisenberg R. J., Harrison S. C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. [PubMed: 16840698]
- Herold B. C., WuDunn D., Soltys N., Spear P. G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991;65:1090–1098. [PMC free article: PMC239874] [PubMed: 1847438]
- Highlander S. L., Dorney D. J., Gage P. J., et al. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J. Virol. 1989;63:730–738. [PMC free article: PMC247744] [PubMed: 2463380]
- Hutchinson L., Johnson D. C. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 1995;69:5401–5413. [PMC free article: PMC189384] [PubMed: 7636985]
- Hutchinson L., Browne H., Wargent V., et al. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 1992a;66:2240–2250. [PMC free article: PMC289017] [PubMed: 1312629]
- Hutchinson L., Goldsmith K., Snoddy D., Ghosh H., Graham F. L., Johnson D. C. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 1992b;66:5603–5609. [PMC free article: PMC289123] [PubMed: 1323714]
- Jogger C. R., Montgomery R. I., Spear P. G. Effects of linker-insertion mutations in herpes simplex virus 1 gD on glycoprotein-induced fusion with cells expressing HVEM or nectin-1. Virology. 2004;318:318–326. [PubMed: 14972557]
- Johnson D. C., Burke R. L., Gregory T. Soluble forms of herpes simplex virus glycoprotein D bind to a limited number of cell surface receptors and inhibit virus entry into cells. J. Virol. 1990;64:2569–2576. [PMC free article: PMC249433] [PubMed: 2159532]
- Johnson R. M., Spear P. G. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 1989;63:819–827. [PMC free article: PMC247755] [PubMed: 2536105]
- Jones N. A., Geraghty R. J. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology. 2004;324:213–228. [PubMed: 15183068]
- Kaye J. F., Gompels U. A., Minson A. C. Glycoprotein H of human cytomegalovirus (HCMV) forms a stable complex with the HCMV UL115 gene product. J. Gen. Virol. 1992;73(10):2693–2698. [PubMed: 1328481]
- Keller P. M., Neff B. J., Ellis R. W. Three major glycoprotein genes of varicella-zoster virus whose products have neutralization epitopes. J. Virol. 1984;52:293–297. [PMC free article: PMC254520] [PubMed: 6207311]
- Kenyon T. K., Cohen J. I., Grose C. Phosphorylation by the varicella-zoster virus ORF47 protein serine kinase determines whether endocytosed viral gE traffics to the trans-Golgi network or recycles to the cell membrane. J. Virol. 2002;76:10980–10993. [PMC free article: PMC136633] [PubMed: 12368341]
- Kousoulas K. G., Huo B., Pereira L. Antibody-resistant mutations in cross-reactive and type-specific epitopes of herpes simplex virus 1 glycoprotein B map in separate domains. Virology. 1988;166:423–431. [PubMed: 2459843]
- Krummenacher C., Nicola A. V., Whitbeck J. C., et al. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 1998;72:7064–7074. [PMC free article: PMC109927] [PubMed: 9696799]
- Krummenacher C., Rux A. H., Whitbeck J. C., et al. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity, while the third domain is involved in oligomerization of HveC. J. Virol. 1999;73:8127–8137. [PMC free article: PMC112829] [PubMed: 10482562]
- Krummenacher C., Baribaud I., Ponce De Leon M., et al. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 2000;74:10863–10872. [PMC free article: PMC113165] [PubMed: 11069980]
- Krummenacher C., Baribaud I., Ponce De Leon M., et al. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology. 2004;322:286–299. [PubMed: 15110526]
- Krummenacher C., Supekar V. M., Whitbeck J. C., et al. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. [PMC free article: PMC1356314] [PubMed: 16292345]
- Kwon B. S., Tan K. B., Ni J., et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 1997;272:14272–14276. [PubMed: 9162061]
- La S., Kim J., Kwon B. S., Kwon B. Herpes simplex virus type 1 glycoprotein D inhibits T-cell proliferation. Mol. Cells. 2002;14:398–403. [PubMed: 12521303]
- Ligas M. W., Johnson D. C. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 1988;62:1486–1494. [PMC free article: PMC253172] [PubMed: 2833603]
- Linehan M. M., Richman S., Krummenacher C., Eisenberg R. J., Cohen G. H., Iwasaki A. In vivo role of nectin-1 in entry of herpes simplex virus type 1 (HSV-1) and HSV-2 through the vaginal mucosa. J. Virol. 2004;78:2530–2536. [PMC free article: PMC369262] [PubMed: 14963155]
- Liu D. X., Gompels U. A., Tomasi L., Fiume G., Foà-, and Campadelli-1993Human herpesvirus-6 glycoprotein H and L homologs are components of the gp100 complex and the gH external domain is the target for neutralizing monoclonal antibodies Virology 19712–22. [PubMed: 7692666]
- Lopez M., Cocchi F., Menotti L., Avitabile E., Dubreuil P., Fiume G., and Campadelli-2000Nectin2a (PRR2a or HveB) and nectin2d are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D J. Virol. 741267–1274. [PMC free article: PMC111461] [PubMed: 10627537]
- Lopez M., Cocchi F., Avitabile E., et al. Novel, soluble isoform of the herpes simplex virus (HSV) receptor nectin1 (or PRR1-HIgR-HveC) modulates positively and negatively susceptibility to HSV infection. J. Virol. 2001;75:5684–5691. [PMC free article: PMC114282] [PubMed: 11356977]
- Mabit H., Nakano M. Y., Prank U., et al. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 2002;76:9962–9971. [PMC free article: PMC136514] [PubMed: 12208972]
- Mallory S., Sommer M., Arvin A. M. Mutational analysis of the role of glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E conformation and trafficking. J. Virol. 1997;71:8279–8288. [PMC free article: PMC192286] [PubMed: 9343180]
- Manoj S., Jogger C. R., Myscofski D., Yoon M., Spear P. G. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl Acad. Sci. USA. 2004;101:12414–12421. [PMC free article: PMC515077] [PubMed: 15273289]
- Manservigi R., Spear P. G., Buchan A. Cell fusion induced by herpes simplex virus is promoted and suppressed by different viral glycoproteins. Proc. Natl Acad. Sci. USA. 1977;74:3913–3917. [PMC free article: PMC431783] [PubMed: 198812]
- Maresova L., Pasieka T. J., Grose C. Varicella-zoster Virus gB and gE coexpression, but not gB or gE alone, leads to abundant fusion and syncytium formation equivalent to those from gH and gL coexpression. J. Virol. 2001;75:9483–9492. [PMC free article: PMC114515] [PubMed: 11533210]
- Marozin S., Prank U., Sodeik B. Herpes simplex virus type 1 infection of polarized epithelial cells requires microtubules and access to receptors at cell–cell contact sites. J. Gen. Virol. 2004;85:775–786. [PubMed: 15039520]
- Martinez W. M., Spear P. G. Amino acid substitutions in the V domain of nectin-1 (HveC) that impair entry activity for herpes simplex virus types 1 and 2 but not for Pseudorabies virus or bovine herpesvirus 1. J. Virol. 2002;76:7255–7262. [PMC free article: PMC136344] [PubMed: 12072525]
- Mata M., Zhang M., Hu X., Fink D. J. HveC (nectin-1) is expressed at high levels in sensory neurons, but not in motor neurons, of the rat peripheral nervous system. J. Neurovirol. 2001;7:476–480. [PubMed: 11582520]
- Matsushima H., Utani A., Endo H., et al. The expression of nectin-1alpha in normal human skin and various skin tumours. Br. J. Dermatol. 2003;148:755–762. [PubMed: 12752135]
- Mauri D. N., Ebner R., Montgomery R. I., et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. [PubMed: 9462508]
- McGeoch D. J., Dalrymple M. A., Davison A. J., et al. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988;69:1531–1574. [PubMed: 2839594]
- Melancon J. M., Foster T. P., Kousoulas K. G. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 2004;78:7329–7343. [PMC free article: PMC434089] [PubMed: 15220406]
- Menotti L., Lopez M., Avitabile E., et al. The murine homolog of human-Nectin1d serves as a species non-specific mediator for entry of human and animal aherpesviruses in a pathway independent of a detectable binding to gD. Proc. Natl Acad. Sci. USA. 2000;97:4867–4872. [PMC free article: PMC18324] [PubMed: 10781093]
- Menotti L., Avitabile E., Dubreuil P., Lopez M., Fiume G., and Campadelli-2001Comparison of murine and human nectin1 binding to herpes simplex virus glycoprotein D (gD) reveals a weak interaction of murine nectin1 to gD and a gD-dependent pathway of entry Virology 282256–266. [PubMed: 11289808]
- Menotti L., Casadio R., Bertucci C., Lopez M., Fiume G., and Campadelli-2002aSubstitution in the murine nectin1 receptor of a single conserved amino acid at a position distal from herpes simplex virus gD binding site confers high affinity binding to gD J. Virol. 765463–5471. [PMC free article: PMC137010] [PubMed: 11991974]
- Menotti L., Cocchi F., Fiume G., and Campadelli-2002bCritical residues in the CC′ ridge of the human nectin1 receptor V domain enable herpes simplex virus entry into the cell and act synergistically with the downstream region Virology 3016–12. [PubMed: 12359441]
- Milne R. S., Connolly S. A., Krummenacher C., Eisenberg R. J., Cohen G. H. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology. 2001;281:315–328. [PubMed: 11277703]
- Milne R. S., Hanna S. L., Rux A. H., Willis S. H., Cohen G. H., Eisenberg R. J. Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J. Virol. 2003;77:8962–8972. [PMC free article: PMC167229] [PubMed: 12885913]
- Mo C., Lee J., Sommer M., Grose C., Arvin A. M. The requirement of varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology. 2002;304:176–186. [PubMed: 12504560]
- Montalvo E. A., Grose C. Neutralization epitope of varicella zoster virus on native viral glycoprotein gp118 (VZV glycoprotein gpⅢ). Virology. 1986;149:230–241. [PubMed: 2418586]
- Montgomery R. I., Warner M. S., Lum B. J., Spear P. G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. [PubMed: 8898196]
- Nicola A. V., Straus S. E. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 2004;78:7508–7517. [PMC free article: PMC434080] [PubMed: 15220424]
- Nicola A. V., Peng C., Lou H., Cohen G. H., Eisenberg R. J. Antigenic structure of soluble herpes simplex virus (HSV) glycoprotein D correlates with inhibition of HSV infection. J. Virol. 1997;71:2940–2946. [PMC free article: PMC191422] [PubMed: 9060653]
- Nicola A. V., McEvoy A. M., Straus S. E. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003;77:5324–5332. [PMC free article: PMC153978] [PubMed: 12692234]
- Ojala P. M., Sodeik B., Ebersold M. W., Kutay U., Helenius A. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell Biol. 2000;20:4922–4931. [PMC free article: PMC85943] [PubMed: 10848617]
- Olson J. K., Grose C. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 1997;71:4042–4054. [PMC free article: PMC191557] [PubMed: 9094682]
- Olson J. K., Grose C. Complex formation facilitates endocytosis of the varicella-zoster virus gE:gI Fc receptor. J. Virol. 1998;72:1542–1551. [PMC free article: PMC124636] [PubMed: 9445058]
- Olson J. K., Santos R. A., Grose C. 1998Varicella-zoster virus glycoprotein gE: endocytosis and trafficking of the Fc receptor J. Infect. Dis. 178 Suppl 1, S2–S6. [PubMed: 9852964]
- Pasieka T. J., Maresova L., Grose C. A functional YNKI motif in the short cytoplasmic tail of varicella-zoster virus glycoprotein gH mediates clathrin-dependent and antibody-independent endocytosis. J. Virol. 2003;77:4191–4204. [PMC free article: PMC150655] [PubMed: 12634377]
- Pasieka T. J., Maresova L., Shiraki K., Grose C. Regulation of varicella-zoster virus-induced cell-to-cell fusion by the endocytosis-competent glycoproteins gH and gE. J. Virol. 2004;78:2884–2896. [PMC free article: PMC353742] [PubMed: 14990707]
- Patera A., Ali M. A., Tyring S., et al. Polymorphisms in the genes for herpesvirus entry. J. Infect. Dis. 2002;186:444–445. [PubMed: 12134247]
- Pellett P. E., Kousoulas K. G., Pereira L., Roizman B. Anatomy of the herpes simplex virus 1 strain F glycoprotein B gene: primary sequence and predicted protein structure of the wild type and of monoclonal antibody-resistant mutants. J. Virol. 1985;53:243–253. [PMC free article: PMC255021] [PubMed: 2981343]
- Peng T., Ponce de Leon M., Novotny M. J., et al. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J. Virol. 1998;72:6092–6103. [PMC free article: PMC110415] [PubMed: 9621073]
- Pereira L., Ali M., Kousoulas K., Huo B., Banks T. Domain structure of herpes simplex virus 1 glycoprotein B: neutralizing epitopes map in regions of continuous and discontinuous residues. Virology. 1989;172:11–24. [PubMed: 2475970]
- Qadri I., Gimeno C., Navarro D., Pereira L. Mutations in conformation-dependent domains of herpes simplex virus 1 glycoprotein B affect the antigenic properties, dimerization, and transport of the molecule. Virology. 1991;180:135–152. [PubMed: 1701945]
- Reymond N., Borg J., Lecocq E., et al. Human nectin3/PRR3: a novel member of the PVR/PRR/nectin family that interacts with afadin. Gene. 2000;255:347–355. [PubMed: 11024295]
- Richart S., Simpson S., Krummenacher C., et al. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 2003;77:3307–3311. [PMC free article: PMC149788] [PubMed: 12584355]
- Roberts S. R., Ponce de Leon M., Cohen G. H., Eisenberg R. J. Analysis of the intracellular maturation of the herpes simplex virus type 1 glycoprotein gH in infected and transfected cells. Virology. 1991;184:609–624. [PubMed: 1653491]
- Roche S., Bressanelli S., Rey F. A., Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. [PubMed: 16840692]
- Roop C., Hutchinson L., Johnson D. C. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 1993;67:2285–2297. [PMC free article: PMC240370] [PubMed: 8383241]
- Rux A. H., Lou H., Lambris J. D., Friedman H. M., Eisenberg R. J., Cohen G. H. Kinetic analysis of glycoprotein C of herpes simplex virus types 1 and 2 binding to heparin, heparan sulfate, and complement component C3b. Virology. 2002;294:324–332. [PubMed: 12009874]
- Santos R. A., Padilla J. A., Hatfield C., Grose C. Antigenic variation of varicella zoster virus Fc receptor gE: loss of a major B cell epitope in the ectodomain. Virology. 1998;249:21–31. [PubMed: 9740773]
- Santos R. A., Hatfield C. C., Cole N. L., et al. Varicella-zoster virus gE escape mutant VZV-MSP exhibits an accelerated cell-to-cell spread phenotype in both infected cell cultures and SCID-hu mice. Virology. 2000;275:306–317. [PubMed: 10998331]
- Schelhaas M., Jansen M., Haase I., Morsdorf D., and Knebel-2003Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells J. Gen. Virol. 842473–2484. [PubMed: 12917468]
- Shukla D., Spear P. G. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 2001;108:503–510. [PMC free article: PMC209412] [PubMed: 11518721]
- Shukla D., Liu J., Blaiklock P., et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. [PubMed: 10520990]
- Shukla D., Canto, Rowe C. L., Spear P. G. Striking Similarity of murine nectin-1alpha to human nectin-1alpha (HveC) in sequence and activity as a glycoprotein D receptor for alphaherpesvirus entry. J. Virol. 2000;74:11773–11781. [PMC free article: PMC112460] [PubMed: 11090177]
- Smith G. A., Gross S. P., Enquist L. W. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl Acad. Sci. USA. 2001;98:3466–3470. [PMC free article: PMC30676] [PubMed: 11248101]
- Sodeik B. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 2000;8:465–472. [PubMed: 11044681]
- Sodeik B., Ebersold M. W., Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997;136:1007–1021. [PMC free article: PMC2132479] [PubMed: 9060466]
- Spear P. G., Shieh M. T., Herold B. C., WuDunn D., Koshy T. I. Heparan sulfate glycosaminoglycans as primary cell surface receptors for herpes simplex virus. Adv. Exp. Med. Biol. 1992;313:341–353. [PubMed: 1332443]
- Spear P. G., Eisenberg R. J., Cohen G. H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. [PubMed: 11017782]
- Struyf F., Posavad C. M., Keyaerts E., Ranst, Corey L., Spear P. G. Search for polymorphisms in the genes for herpesvirus entry mediator, nectin-1, and nectin-2 in immune seronegative individuals. J. Infect. Dis. 2002;185:36–44. [PubMed: 11756979]
- Takai Y., Irie K., Shimizu K., Sakisaka T., Ikeda W. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci. 2003;94:655–667. [PubMed: 12901789]
- Singer R., Peng C., Ponce De Leon M., et al. Tal-1995Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules J. Virol. 694471–4483. [PMC free article: PMC189189] [PubMed: 7769707]
- Turner A., Bruun B., Minson T., Browne H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998;72:873–875. [PMC free article: PMC109452] [PubMed: 9420303]
- Ward P. L., Fiume G., Avitabile E., Roizman B., Campadelli-1994Localization and putative function of the UL20 membrane protein in cells infected with herpes simplex virus 1 J. Virol. 687406–7417. [PMC free article: PMC237183] [PubMed: 7933124]
- Warner M. S., Geraghty R. J., Martinez W. M., et al. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology. 1998;246:179–189. [PubMed: 9657005]
- Whitbeck J. C., Muggeridge M. I., Rux A. H., et al. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J. Virol. 1999;73:9879–9890. [PMC free article: PMC113037] [PubMed: 10559300]
- Willis S. H., Rux A. H., Peng C., et al. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 1998;72:5937–5947. [PMC free article: PMC110398] [PubMed: 9621056]
- WuDunn D., Spear P. G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989;63:52–58. [PMC free article: PMC247656] [PubMed: 2535752]
- Yoon M., Zago A., Shukla D., Spear P. G. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 2003;77:9221–9231. [PMC free article: PMC187404] [PubMed: 12915538]
- Zhou G., Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc. Natl Acad. Sci. USA. 2006;103:5508–5513. [PMC free article: PMC1459385] [PubMed: 16554374]
- Zhou G., Avitabile E., Fiume G., Roizman B., Campadelli-2003The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell-cell fusion and binding to nectin1 J. Virol. 773759–3767. [PMC free article: PMC149540] [PubMed: 12610150]
Figures
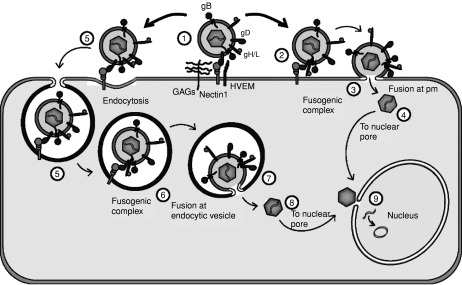
Fig. 7.1
Schematic drawing of HSV entry. Entry can occur either by endocytosis (pathway at the left), or by fusion at the plasma membrane (pathway at the right). Following attachment to cells, gD binds to a cellular receptor (frame 1), and presumably following a conformational change it recruits the glycoproteins B, H and L in an active fusogenic complex (frame 2), triggering the fusion between viral envelope and cellular plasma membrane (frame 3). The naked nucleocapsids are transported to the nucleus (frames 4 and 9). In a cell line-dependent manner or with modified forms of the receptor (see text for details) bound virions can enter cells by endocytosis (frame 5). It is conceivable that at this stage the four fusogenic glycoproteins are in a non-fusion-active form. Following acidification/maturation of the endocytic vesicles, a fusogenic complex may form (frame 6) and fusion ensue between the virion envelope and the vesicle membrane (frame 7). Nucleocapsids delivered to the cytoplasm are transported to the nucleus (frames 8 and 9).

Fig. 7.2
Scanning electron micrographs of HSV -1 and VZV. Images of both viruses were taken by SEM after infection of cultured cells. HSV -1 virions have a more uniform appearance although indentations are seen in an occasional virion envelope (a). In contrast, VZV virions are more aberrant (b). Several of the virions have indentations, while other virions have incomplete envelopes. Since both viruses were examined under the same SEM conditions, it is unlikely that the aberrant nature of the VZV envelope is due to fixation artifacts. Micrographs kindly provided by Dr. Charles Grose.
%20bound%20to%20HVEM%20receptor%20(HveA%2C%20colored%20in%20green)%20as%20determined%20by%20X-ray%20crystallography.&p=BOOKS&id=47385_9780521827140c07_fig003.jpg "Click on image to zoom")
Fig. 7.3
Ribbon diagram of the 3D structure of a soluble truncated form of gD (gD285t, colored in orange) bound to HVEM receptor (HveA, colored in green) as determined by X-ray crystallography. The N-terminus (residues 1–37) of gD is devoid of a specific structure when in the unbound state, but folds into a hairpin when bound to HVEM receptor. The β-strand formed by residues 27–29 (indicated with number 1) forms an intermolecular β-sheet with HVEM residues 35–37 (letter d). The core of gD (residues 56–184) has a V-type immunglobulin domain structure, composed of 9 parallel and antiparallel β-strands (letters A to G) that form two opposing β-sheets, and carries an additional α-helix (α1). The residues 185–259 form two α-helices that fold back to the N-ter (α2 and α3), and two β-strands (numbered 3 and 4). The α3 helix supports gD’s N-terminal hairpin. An additional β-strand (number 2) is located in the connector sequence (residues 33–55) that precedes the Ig-like core. Reprinted from (Carfi et al., 2001), with permission. (See color plate section.)
Tables
Table 7.1Human HSV receptors and the viral strains for which they serve
| Receptor name | Alternative name | Human α-herpesvirus | ||||
|---|---|---|---|---|---|---|
| Unrestricted/rid | Animal α-herpesvirus | |||||
| HSV-1 | HSV-2 | mutants | PrV | BHV-1 | ||
| HVEM | HveA | + | + | − | − | − |
| Nectin1 | PRR1, HI gR, HveC | + | + | + | + | + |
| Nectin2 | PRR2, HveB | − | +/− | + | + | − |