NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Riegert-Johnson DL, Boardman LA, Hefferon T, et al., editors. Cancer Syndromes [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2009-.
Introduction
Birt-Hogg-Dubé (BHD) syndrome is a rare, autosomal dominantly inherited genodermatosis characterized by multiple cutaneous hamartomas (namely fibrofolliculomas and trichodiscomas) and increased susceptibility to renal neoplasms, pulmonary cysts, and spontaneous pneumothoraces. It is caused by mutations in the FLCN gene, which encodes a tumor-suppressor protein, folliculin.
Epidemiology
Birt-Hogg-Dubé (BHD) syndrome is rare. There are no high-quality studies estimating the incidence or prevalence of the disease. It is thought to be underdiagnosed (1).
Historical Background
In 1925, Burnier and Rejsek described a 56-year-old female with multiple, small, skin-colored papules on the head and neck. Their report is probably the first case of what is now called BHD. They termed the histopathological findings perifollicular fibromas (2). Burnier and Rejsek’s report can be read by linking to “The original description of perifollicular fibromas (Birt-Hogg-Dubé syndrome.)”
In 1960, an additional five cases with clinical and histopathologic features similar to Burnier and Rejsek were reported by Zackheim and Pinkus (3). Many similar reports followed. In 1974, Pinkus described three adult patients with widely distributed, asymptomatic, smooth, skin-colored papules. These lesions were named trichodiscomas and were described as “[having] constant topographical relation to hair follicles, unusual complex composition of mucinous and richly vascularized dermal tissue containing both collagen and elastic fibers, presence of small melanized cells in the substance of the tumor, and occasional entry of a thick, myelinated nerve into the lesion” (4). In none of the above described reports were associated systemic manifestations of BHD described.
In 1975, Hornstein and Knickerberg reported on a 47-year-old woman and her 42-year-old brother with multiple perifollicular fibromas. The female patient also had multiple adenomatous colon polyps, one with malignant transformation. The father of these siblings had kidney and lung cysts with severe hemorrhoid complaints. This was thought to represent a new entity separate from Gardner syndrome (5).
During an investigation of a family with hereditary medullary carcinoma of the thyroid, it was noted by Drs. Birt, Hogg, and Dubé that a portion of patients had skin lesions that had not previously been described in association with thyroid cancer. Within a sibship of nine, six members had thyroid cancer. Two members with thyroid cancer and two without had skin lesions described as multiple, small, dome-shaped, yellow- to skin-colored papules measuring 2-4 mm in diameter. They were located most commonly on the head and neck. They also noted acrochordon-like lesions admixed with these. Among the kindred, only patients older than 25 years of age had the above described skin lesions. Histopathologic review of biopsy specimens revealed anastomosing strands of epithelium within a well-circumscribed background of fibrous tissue proliferating outward from a central hair follicle. These lesions were thought to be similar to the lesions described by Rejsek, and they were termed fibrofolliculomas. While fibrofolliculomas were seen in ten lesions, trichodiscomas were seen in four. The acrochordons had the typical appearance upon histopathologic examination (6). The triad of fibrofolliculomas, trichodiscomas, and acrochordons later became known as Birt-Hogg-Dubé syndrome.
Multiple systemic associations have been described in patients with BHD syndrome. In 1986, the first case of fibrofolliculomas, colon polyps, and spontaneous pneumothorax was detailed (7). In 1993, the first case of renal cell carcinoma in a patient with hundreds of fibrofolliculomas was described. The patient’s mother had multiple skin tags, but no history of renal cancer (8). Other conditions reported in patients with Birt-Hogg-Dubé syndrome have included intestinal polyposis (9, 10), connective tissue nevus (11, 12), oral lesions (11, 13), lipomas, angiolipomas, parathyroid adenomas (14), flecked chorioretinopathy (15), parotid oncocytoma (16), multinodular goiter (17), neurothekoma, meningioma (18), and facial angiofibromas (19). Since the initial description of thyroid malignancy associated with BHD syndrome, subsequent studies have failed to demonstrate this particular relationship. It was later discovered that the initially described cohort had also acquired dominantly inherited multiple endocrine neoplasia type 2 (11).
In 2001, the BHD gene locus was mapped using linkage analysis to chromosome 17p11.2 (20). One year later, germline insertion/deletion and nonsense mutations were identified in the FLCN gene at this locus (21).
Pathophysiology
Folliculin
Other hamartomatous syndromes that are associated with increased risk of malignancy (Cowden syndrome, tuberous sclerosis, Peutz-Jeghers syndrome) share a common pathogenesis through impaired control of the mammalian target of rapamycin (mTOR) pathway. This signaling pathway regulates cell growth, and it was hypothesized that BHD may also be associated with dysregulation of this pathway (22).
A 130 kDa protein termed FLCN-interacting protein, FNIP1, interacts with the C terminal of folliculin. The region of folliculin where FNIP1 interacts is the region where nearly all BHD mutations are predicted to result in truncation. 5’ AMP-activated protein kinase (AMPK) is a molecule that negatively regulates mTOR activity. FNIP1 was shown to be phosphorylated by AMPK. Inhibition of AMPK led to reduced FNIP1 expression and reduced FLCN phosphorylation. This implicates folliculin and FNIP1 in the AMPK/mTOR signaling pathway, possibly through energy or nutrient sensing (22).
In schizosaccaromyces pombe yeast, tuberous sclerosis (TSC) proteins and folliculin have opposing roles in regulation of amino-acid homeostasis along common signaling pathways. TSC proteins hamartin and tuberin inhibit the activity of the mammalian target of rapamycin complex 1 (TORC1). A mouse model was developed with inactivation of the BHD gene. The mice developed renal cysts and tumors similar to those of BHD patients. The cysts and tumors demonstrated reduced levels of phosphorylation of ribosomal protein S6, indicating low mTOR activity (23).
Folliculin Genetics
The FLCN gene encodes two alternatively spliced transcripts (Figure 1a and Figure 1b). Isoform 1 spans 25 kb genomic sequence and generates a 3723-bp transcript encoding a 64-KDa protein of 579 amino acids. Isoform 2 encodes 8 exons and a shorter protein of 342 amino acids. The first three exons in both transcripts do not code for protein and are not conserved at the DNA level (24, 25).
Animal models
Nihon rat
A hereditary renal carcinoma rat model was identified in the Sprague-Dawley rat by Japanese investigators and termed the Nihon rat (26). The animals develop multiple renal cysts inherited in a dominant fashion. Histologic examination of these lesions revealed atypical hyperplasia of the renal tubules with clear cell and mixed cell adenomas and carcinomas. Later, the gene was mapped to rat chromosome 10, homologous to human chromosome 17p11.2. This was identified as the rat BHD homologue and a germline premature stop codon mutation was identified.
Heterozygote rats for this mutation have numerous renal tubular tumors at 8 weeks of age, with cellular atypia as early as 4 weeks of age. These lesions later develop into clear cell and mixed cell adenomas and carcinomas. Twenty percent of heterozygotes die or become extremely ill with severe anemia or renal failure before one year of age (26-28). The skin and pulmonary findings seen in human patients with BHD are not seen. Other associated findings included clear cell hyperplasia of endometrium and salivary gland, and rhabdomyolysis.
Homozygotes die early in fetal life.
Other animal models
German shepherd dogs have been noted to develop a naturally occurring kidney cancer syndrome termed hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis (RCND). These canines have bilateral, multifocal renal tumors and numerous firm skin nodules. Females develop uterine leiomyomas. A disease-associated mutation was identified within the canine FLCN homologue (29).
A drosophila BHD homologue has also been described. A decreased gene expression of BHD in this model led to dysregulation of signaling pathways involved in male germline stem cell maintenance (30).
A kidney-specific BHD knockout mouse model was reported in 2008. BHDflox/flox/Ksp-Cre knockout mice developed renal enlargement with polycystic kidneys, hyperplasia, and cystic renal cell carcinoma. Affected mice died at 3 weeks of age from renal failure with blood urea nitrogen levels ten times higher than controls. Mice that were homozygous for the BHD mutation did not survive past 3-8 days post conception. Inactivation of BHD in BHDflox/flox/Ksp-Cre knockout mice led to activation of mTOR. Administration of rapamycin (an mTOR inhibitor) to these mice prolonged their survival (31).
Dermatologic Manifestations
Dermatologic findings may be the presenting signs in patients with BHD syndrome; therefore, it is important to recognize these so that further systemic evaluation can be undertaken. Clinical photographs are shown in Figures 2, 3, 4, 5, 6, 7, 8, 9, and 10. Skin findings usually do not appear before the patient reaches 20 years of age. The characteristic skin lesions are described as multiple, 2-4 mm, waxy, white, opaque, smooth, dome-shaped papules most commonly involving the head and neck, with less frequent involvement of the trunk. Biopsy of these lesions shows fibrofolliculoma or less commonly trichodiscoma. It is not possible to distinguish fibrofolliculomas from trichodiscomas on clinical grounds. Acrochordon-like lesions have also been noted, localized to the eyelids, axillae, and skin folds (Figures 4 and 5). Biopsy of these lesions may show fibrofolliculoma or simple fibroepitheliomatous polyp features. Some may have comedo- or cyst-like features (6). Some patients manifest only a few lesions in a localized distribution, while others have hundreds in a generalized distribution. The areas on the face most often involved are the nose, then cheeks, forehead, and ears. Oral examination is important, as these patients may have multiple, discrete, soft, papules of the lips, gingival, and buccal mucosa (11, 13). Other associated skin findings have included connective tissue nevus (11, 12), lipomas, angiolipomas (14), and facial angiofibromas (19).
The differential diagnosis for the multiple facial papules seen in BHD includes several familial cancer syndromes. While punch biopsy is necessary to establish a definitive diagnosis, there are distinguishing clinical features. Fibrofolliculomas and trichodiscomas appear as described above. Multiple trichilemmomas are seen in Cowden syndrome. These lesions look similar to fibrofolliculomas and are located around the mouth, nose, and ears. Associated findings include cobblestoning of the oral mucosa with keratoses at acral sites. Multiple trichoepitheliomas are seen in Brooke-Spiegler syndrome. These lesions are larger, 2-8 mm, firm papules and are located on the nasolabial folds, forehead, upper lip, and scalp. Multiple angiofibromas are seen in tuberous sclerosis. These lesions are pink, vascular-appearing papules and are located periorally. Associated findings include periungual fibromas (32).
There have been case reports of BHD syndrome patients with basal cell carcinoma, and one patient has been reported with malignant melanoma (8, 11). Another patient has been reported with a family history of malignant melanoma (20).
There are limited reports addressing treatment of fibrofolliculomas and trichodiscomas. Excision is often unreasonable given the large numbers of lesions. Electrocautery, curettage, and ablative laser modalities have been used with varied success. While the lesions may resolve post-treatment, they will often recur (33-35).
Dermatopathology
BHD syndrome is characterized by the triad of fibrofolliculomas, trichodiscomas, and acrochordons. Fibrofolliculomas and trichodiscomas are benign hamartomatous growths with similar histologic features (Figures 11, 12, 13, and 14). In fact, some have suggested that the slight differences in histologic pattern may represent a spectrum of the same mesodermal proliferation (36-38). Multiple deeper sections may reveal features of both fibrofolliculoma and trichodiscoma within the same lesion.
Fibrofolliculoma
Fibrofolliculomas are derived from both hair follicle epithelium and mesenchyme (Figure 11). The hair follicle is well formed and may be dilated and filled with keratinaceous debris. The peri-infundibular area is ensheathed by a well-circumscribed proliferation of fibrous tissue containing collagen and hyaluronic acid. Anastomosing 2-4–cell thick epithelial strands proliferate outward from the follicular infundibulum. Unlike trichofolliculomas, these strands do not terminate with hair follicle structures (6).
Trichodiscoma
Trichodiscomas are derived principally from pilosebaceous mesenchyme (Figure 12). The bulk of the lesion is composed of fibrous stroma with overlying attenuated epidermis in close proximity to a hair follicle. There is a collarette involving the lateral aspect of the lesion, while the central epidermis is flattened (4).
Acrochordon
This lesion may have features of a fibroepitheliomatous polyp or may show localized features of fibrofolliculoma (6). Because of this, some authors do not include this as an independent component of BHD syndrome. In addition, skin tags are extremely common in adult patients, thus making it difficult to establish them as a marker for a genetic syndrome (36).
Renal Manifestations
Patients with BHD syndrome have a 20-30% lifetime risk of developing one or more renal tumors (39). While sporadic renal cancer occurs predominantly in men, renal cancer in BHD syndrome occurs equally in both genders, as in other hereditary renal cancer syndromes (40).
The mean age of presentation of renal cancer in BHD syndrome is 50 years, although this can range from as early as age 20 to as late as age 70 (41, 42). In contrast, most cases of sporadic renal cancer develop in between the sixth and seventh decade of life. There have also been several reports of BHD patients with multiple renal cysts, often occurring after age 40 (1, 5, 20, 43-45). It has been hypothesized that multiple renal cysts may represent a manifestation of BHD syndrome, particularly in BHD syndrome families with no detectable mutation in FLCN. Ultrasound, computated tomography (CT) and magnetic resonance imaging (MRI) can all be used to evaluate and diagnose renal tumors in patients with BHD (Figures 15 and 16.)
BHD syndrome differs from other hereditary renal cancer syndromes by the diversity of histological subtypes. FLCN is considered to act as a tumor suppressor gene in the kidneys (21), and different renal tumors in one BHD patient are caused by distinct second hits, i.e., somatic mutations or loss of heterozygosity of the FLCN gene (46). The most common and characteristic tumor types in BHD syndrome are hybrid chromophobe/oncocytic (50%) and chromophobe (33%) renal cancer (Figures 17, 18, and 19). Less commonly, oncocytoma (5%), clear cell carcinomas (9%), and papillary carcinomas can occur (47). The tumors may be solitary and unilateral or multifocal and bilateral.
When renal cancer is diagnosed in a patient with BHD syndrome, kidney sparing surgery should be considered as in other hereditary types of renal cancer (48). Anesthesiologists should be aware of the potential for perioperative pneumothorax in these patients.
Pulmonary Manifestations
Approximately 25% of BHD patients will experience pneumothorax which will typically occur during third to sixth decades of life (49, 50). Patients with BHD syndrome who present with cystic lung disease or a history of pneumothoraces tend to be middle-aged without a previous diagnosis of the underling genetic syndrome (Figures 20, 21, 22) (49, 50). Approximately 90% of adult patients with BHD will have lung cysts of varying degree on CT (Movie clip 1) (51). Some patients can have pulmonary manifestations (cystic lung diseased and pneumothorax) in the absence of BHD skin or kidney lesions (51). Histopathologic features in the lung are nonspecific and include intraparenchymal air-filled spaces surrounded by normal parenchyma or a thin fibrous wall (49).
Cystic lung disease seen in BHD syndrome needs to be distinguished from other lung diseases characterized by multifocal or diffuse cystic changes, including lymphangioleiomyomatosis (LAM), pulmonary Langerhans’ cell histiocytosis (PLCH), lymphocytic interstitial pneumonitis (LIP), and Pneumocystis pneumonia (52). Pulmonary lymphangioleiomyomatosis, whether occurring in a sporadic form or associated with tuberous sclerosis complex (TSC), affects women of childbearing age almost exclusively but can be associated with renal tumors (angiomyolipomas) and may at times be difficult to distinguish from BHD (53). Lymphocytic interstitial pneumonia and Pneumocystis pneumonia both cause parenchymal changes aside from cysts such as ground-glass opacities, consolidation, nodules, and reticular opacities (52). In addition, both of these disorders are usually symptomatic and are associated with specific clinical contexts. Pulmonary Langerhans’ cell histiocytosis encountered in adults is usually a smoking-related interstitial lung disease and is characterized by irregular cystic lesions and nodules predominantly affecting the upper and mid lung zones with relative sparing of the bases (54). Rarely, metastatic neoplasms, such as adenocarcinomas and low-grade sarcomas, can present with cystic lung lesions (52).
Diagnosis
The diagnosis is usually made on the basis of the cutaneous features, which appear in the mid-twenties. A few patients have pneumothorax as a presenting sign, and this can occur earlier than age 20. It is important to note that the typical triad of findings may not occur in every patient and that the onset of each is variable. Because there is considerable intra- and inter-familial variance of BHD expression, diagnostic criteria should be inclusive in order to minimize the amount of underdiagnosed cases of BHD. The diagnosis of BHD syndrome should be considered for patients with any of the following:
- Renal cancer of any histologic type before the age of 50 years and/or multiple renal tumors in one individual of any age, plus pneumothorax in two or more relatives or more than one pneumothorax in a single relative.
- Histologically proven diagnosis of fibrofolliculoma in a single patient.
- Hybrid chromophobe/oncocytic renal tumor in a single patient.
- Chromophobe renal tumors in two first-degree relatives.
- Pneumothorax in two first-degree relatives or in more than two relatives of any degree.
Management
There are no consensus or organization-approved cancer surveillance or management guidelines for patients with BHD. The recommendations below are provisional (Table 1). All patients should be strongly counseled to discontinue smoking, as it is a known risk factor for kidney cancer and lung disease.
Renal cancer
Lifelong renal surveillance should begin at 20 years of age. Although ultrasound is an adequate initial investigation in patients with symptoms suggestive of renal disease, it is less appropriate as a screening method in asymptomatic patients. Sensitivity of ultrasound to visualize tumors with a diameter of 1-3 cm is about 70% and declines with smaller size. The sensitivity of CT in demonstrating small renal tumors is much better. However, the radiation burden accumulates rapidly when performing regular CT scans from an early age, and there is growing concern on the potential for cancer induction (55-57). CT and MRI generally have comparable qualities with regard to cancer detection (58), but MRI has the advantage that no radiation is needed. Thus, MRI may be considered the most appropriate first imaging method in patients with higher risk of developing renal tumors (59, 60). Some have suggested an MRI as an initial means of evaluation, followed by either yearly MRI or ultrasound studies. Others have recommended CT scanning every 3-5 years (45).
Pneumothorax
Patients should be counseled on the symptoms of pneumothorax, including shortness of breath and chest discomfort. For asymptomatic patients, the authors recommend an initial baseline high resolution chest CT with follow-up studies every 3 to 5 years. For symptomatic patients or those with chronic pneumothorax, follow up should be individualized.
Low pressure environments, such as high altitudes or air travel, may slightly increase the risk of a pneumothorax. The risk of pneumothorax after air travel has been best studied for patients with lymphangiolieomyomatosis (LAM). In one study of LAM patients, 1 in 100 patients had a new pneumothorax after air travel (61). If a patient with BHD syndrome has any symptoms of pneumothorax after being at high altitude environment, he or she should immediately seek medical attention.
The treatment of pneumothorax for BHD patients is the same as it is for the general population. The American College of Chest Physicians has developed guidelines for the treatment of pneumothorax which can be seen by clicking on this link. Mainstays of treatment are chest tube drainage and pleurodesis.
Colon and Skin
While there were initial reports of an association between fibrofolliculomas and colon cancer, this association was not confirmed in later reports. Patients with BHD syndrome should undergo routine colon cancer screening as is recommended for the general population. There are reports of BHD patients being diagnosed with melanoma and routine dermatologic evaluation is recommended (45).
Clinical Genetics
Approximately 60-80% of BHD patients have identifiable mutations in the folliculin gene, FLCN. As of 2009, the FLCN database reported 53 unique germline mutations: 45% deletions, 32% substitutions, 15% duplications, 6% insertion/deletions, and 2% insertions (62). The most commonly encountered mutation occurs as a C insertion or deletion in exon 11. It is predicted that mutations in this hotspot cause truncation of the folliculin protein (20, 42).
No confirmed genotype-phenotype correlations have been identified. There is a report that patients with the C deletion mutation in the hypermutable hotspot of the FLCN gene have a significantly lower risk of developing renal tumors (6%) than patients with the C insertion mutation in this hotspot (33%) (47).
There is a limited role for genetic testing for FLCN mutations in BHD patients. A negative test result does not rule out the diagnosis of BHD as 20-40% of patients may have a negative test but still meet clinical diagnostic criteria. Also, FLCN genetic testing results do not change patient management at this time.
A list of laboratories offering FLCN genetic testing is available at www.geneclinics.org.
Movie Clip Captions
High resolution computated tomography (CT) scan of the lungs of a patient with Birt-Hogg-Dubé syndrome. Throughout both lungs there are emphysematous changes and bullae (cysts). Also seen are small ground glass nodules in the left upper lung and changes in the right lung from previous thoracotomy. See also Figures 20, 21, and 22.
References
- 1.
- Lindor N.M. et al. Birt-Hogg-Dube syndrome: an autosomal dominant disorder with predisposition to cancers of the kidney, fibrofolliculomas, and focal cutaneous mucinosis. Int J Dermatol. 2001;40(10):653–6. [PubMed: 11737429]
- 2.
- Rejsek and Burnier, Fibromes sous-cutanes peripilares multiples du cou. Bull Soc francq dermat et syph, 192532: p. 242-243.
- 3.
- Zackheim H.S., Pinkus H. Perifollicular Fibromas. Arch Dermatol. 1960;82:913–919. [PubMed: 13787611]
- 4.
- Pinkus H., Coskey R., Burgess G.H. Trichodiscoma. A benign tumor related to haarscheibe (hair disk). J Invest Dermatol. 1974;63(2):212–8. [PubMed: 4843452]
- 5.
- Hornstein O., Knickenberg M. Perifollicular Fibromatosis Cutis with Polyps of the Colon - a Cutaneo-Intestinal Syndrome sui generis. Arch Derm Res. 1975;253:161–175. [PubMed: 1200700]
- 6.
- Birt A.R., Hogg G.R., Dube J. Hereditary Multiple Fibrofolliculomas With Trichodiscomas and Acrochordons. Arch Dermatol. 1977;113:1674–1677. [PubMed: 596896]
- 7.
- Binet O. et al. Fibromes perifolliculaires polypose colique familaile pneumothorax spontanes familiaux. Ann Dermatol Venereol. 1986;113:928–930.
- 8.
- Roth J.S. et al. Bilateral renal cell carcinoma in the Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 1993;29(6):1055–6. [PubMed: 8245249]
- 9.
- LeGuyadec T. et al. Trichodiscomes Multiples Associes A Une Polypose Colique. Ann Dermatol Venereol. 1998;125:717–719. [PubMed: 9835964]
- 10.
- Rongioletti F. et al. Fibrofolliculomas, tricodiscomas and acrochordons (Birt-Hogg-Dube) associated with intestinal polyposis. Clin Exp Dermatol. 1989;14(1):72–4. [PubMed: 2805394]
- 11.
- Toro J.R. et al. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135(10):1195–202. [PubMed: 10522666]
- 12.
- Weintraub R., Pinkus H. Multiple fibrofolliculomas (Birt-Hogg-Dube) associated with a large connective tissue nevus. J Cutan Pathol. 1977;4(6):289–99. [PubMed: 753849]
- 13.
- Nadershahi N.A., Wescott W.B., Egbert B. Birt-Hogg-Dube syndrome: a review and presentation of the first case with oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83(4):496–500. [PubMed: 9127384]
- 14.
- Chung J.Y. et al. Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol. 1996;35(5):365–7. [PubMed: 8734663]
- 15.
- Walter P. et al. Flecked chorioretinopathy associated with Birt-Hogg-Dube syndrome. Graefes Arch Clin Exp Ophthalmol. 1997;235(6):359–61. [PubMed: 9202964]
- 16.
- Liu V., Kwan T., Page E.H. Parotid oncocytoma in the Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2000;43(6):1120–2. [PubMed: 11100034]
- 17.
- Drummond C., Grigoris I., Dutta B. Birt-Hogg-Dube syndrome and multinodular goitre. Australas J Dermatol. 2002;43(4):301–4. [PubMed: 12423440]
- 18.
- Vincent A. et al. Birt-Hogg-Dube syndrome: two patients with neural tissue tumors. J Am Acad Dermatol. 2003;49(4):717–9. [PubMed: 14512924]
- 19.
- Schaffer J.V. et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53(2) Suppl 1:S108–11. [PubMed: 16021156]
- 20.
- Khoo S.K. et al. Birt-Hogg-Dube syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. 2001;20(37):5239–42. [PubMed: 11526515]
- 21.
- Nickerson M.L. et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2(2):157–64. [PubMed: 12204536]
- 22.
- Baba M. et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103(42):15552–7. [PMC free article: PMC1592464] [PubMed: 17028174]
- 23.
- Hartman T.R. et al. The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28(13):1594–604. [PMC free article: PMC2664853] [PubMed: 19234517]
- 24.
- Hasumi Y. et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. 2009;106(44):18722–7. [PMC free article: PMC2765925] [PubMed: 19850877]
- 25.
- Hudon, V., et al., Renal tumor suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J Med Genet, 2009. [PubMed: 19843504]
- 26.
- Kouchi M. et al. Natural history of the Nihon (Bhd gene mutant) rat, a novel model for human Birt-Hogg-Dube syndrome. Virchows Arch. 2006;448(4):463–71. [PubMed: 16447066]
- 27.
- Okimoto K. et al. A novel "Nihon" rat model of a Mendelian dominantly inherited renal cell carcinoma. Jpn J Cancer Res. 2000;91(11):1096–9. [PMC free article: PMC5926279] [PubMed: 11092972]
- 28.
- Okimoto K. et al. A germ-line insertion in the Birt-Hogg-Dube (BHD) gene gives rise to the Nihon rat model of inherited renal cancer. Proc Natl Acad Sci U S A. 2004;101(7):2023–7. [PMC free article: PMC357045] [PubMed: 14769940]
- 29.
- Lingaas F. et al. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Hum Mol Genet. 2003;12(23):3043–53. [PubMed: 14532326]
- 30.
- Singh S.R. et al. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006;25(44):5933–41. [PubMed: 16636660]
- 31.
- Chen J. et al. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS One. 2008;3(10):e3581. [PMC free article: PMC2570491] [PubMed: 18974783]
- 32.
- Vincent A. et al. Birt-Hogg-Dube syndrome: a review of the literature and the differential diagnosis of firm facial papules. J Am Acad Dermatol. 2003;49(4):698–705. [PubMed: 14512919]
- 33.
- Farrant P.B., Emerson R. Letter: hyfrecation and curettage as a treatment for fibrofolliculomas in Birt-Hogg-Dube syndrome. Dermatol Surg. 2007;33(10):1287–8. [PubMed: 17903168]
- 34.
- Gambichler T. et al. Treatment of Birt-Hogg-Dube syndrome with erbium:YAG laser. J Am Acad Dermatol. 2000;43(5 Pt 1):856–8. [PubMed: 11050594]
- 35.
- Jacob C.I., Dover J.S. Birt-Hogg-Dube syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137(1):98–9. [PubMed: 11176677]
- 36.
- De la Torre C. et al. Acrochordons are not a component of the Birt-Hogg-Dube syndrome: does this syndrome exist? Case reports and review of the literature. Am J Dermatopathol. 1999;21(4):369–74. [PubMed: 10446780]
- 37.
- Fujita W.H., Barr R.J., Headley J.L. Multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1981;117(1):32–5. [PubMed: 7458379]
- 38.
- Collins G.L., Somach S., Morgan M.B. Histomorphologic and immunophenotypic analysis of fibrofolliculomas and trichodiscomas in Birt-Hogg-Dube syndrome and sporadic disease. J Cutan Pathol. 2002;29(9):529–33. [PubMed: 12358810]
- 39.
- Pavlovich C.P. et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. 2002;26(12):1542–52. [PubMed: 12459621]
- 40.
- Toro J.R. et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321–31. [PMC free article: PMC2564862] [PubMed: 18234728]
- 41.
- Khoo S.K. et al. Clinical and genetic studies of Birt-Hogg-Dube syndrome. J Med Genet. 2002;39(12):906–12. [PMC free article: PMC1757219] [PubMed: 12471204]
- 42.
- Schmidt L.S. et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. 2005;76(6):1023–33. [PMC free article: PMC1196440] [PubMed: 15852235]
- 43.
- Gupta P. et al. Radiological findings in Birt-Hogg-Dube syndrome: a rare differential for pulmonary cysts and renal tumors. Clin Imaging. 2007;31(1):40–3. [PubMed: 17189846]
- 44.
- Kluijt I. et al. Early onset of renal cancer in a family with Birt-Hogg-Dube syndrome. Clin Genet. 2009;75(6):537–43. [PubMed: 19320655]
- 45.
- Welsch M.J., Krunic A., Medenica M.M. Birt-Hogg-Dube Syndrome. Int J Dermatol. 2005;44(8):668–73. [PubMed: 16101870]
- 46.
- Vocke C.D. et al. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dube-associated renal tumors. J Natl Cancer Inst. 2005;97(12):931–5. [PubMed: 15956655]
- 47.
- Linehan W.M. et al. Identification of the genes for kidney cancer: opportunity for disease-specific targeted therapeutics. Clin Cancer Res. 2007;13(2 Pt 2):671s–679s. [PubMed: 17255292]
- 48.
- Pavlovich C.P. et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol. 2005;173(5):1482–6. [PubMed: 15821464]
- 49.
- Ayo D.S. et al. Cystic lung disease in Birt-Hogg-Dube syndrome. Chest. 2007;132(2):679–84. [PubMed: 17505035]
- 50.
- Toro J.R. et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube syndrome. Am J Respir Crit Care Med. 2007;175(10):1044–53. [PMC free article: PMC1899269] [PubMed: 17322109]
- 51.
- Gunji Y. et al. Mutations of the Birt Hogg Dube gene in patients with multiple lung cysts and recurrent pneumothorax. J Med Genet. 2007;44(9):588–93. [PMC free article: PMC2597956] [PubMed: 17496196]
- 52.
- Ryu J.H., Swensen S.J. Cystic and cavitary lung diseases: focal and diffuse. Mayo Clin Proc. 2003;78(6):744–52. [PubMed: 12934786]
- 53.
- Ryu J.H. et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–11. [PMC free article: PMC2662978] [PubMed: 16210669]
- 54.
- Vassallo R., Ryu J.H. Pulmonary Langerhans' cell histiocytosis. Clin Chest Med. 2004;25(3):561–71. [PubMed: 15331192]
- 55.
- Brenner D.J. Radiation risks potentially associated with low-dose CT screening of adult smokers for lung cancer. Radiology. 2004;231(2):440–5. [PubMed: 15128988]
- 56.
- Brenner D.J. AJR Am J Roentgenol. 2001;231:440–5.
- 57.
- Wall B.F. et al. What are the risks from medical X-rays and other low dose radiation? Br J Radiol. 2006;79(940):285–94. [PubMed: 16585719]
- 58.
- Coll D.M., Smith R.C. Update on radiological imaging of renal cell carcinoma. BJU Int. 2007;99 5 Pt B:1217–22. [PubMed: 17441914]
- 59.
- Ozsoy, M., et al., Surveillance for the management of small renal masses. Adv Urol, 2008: p. 196701. [PMC free article: PMC2515364] [PubMed: 18704192]
- 60.
- Semelka R.C. et al. Imaging strategies to reduce the risk of radiation in CT studies, including selective substitution with MRI. J Magn Reson Imaging. 2007;25(5):900–9. [PubMed: 17457809]
- 61.
- Taveira-DaSilva A.M. et al. Pneumothorax after air travel in lymphangioleiomyomatosis, idiopathic pulmonary fibrosis, and sarcoidosis. Chest. 2009;136(3):665–70. [PMC free article: PMC2775992] [PubMed: 19318672]
- 62.
- Wei M.H. et al. The folliculin mutation database: an online database of mutations associated with Birt-Hogg-Dube syndrome. Hum Mutat. 2009;30(9):E880–90. [PMC free article: PMC3234166] [PubMed: 19562744]
Figures

Figure 1a. Genomic structure of FLCN1. Main transcript encoding full-length protein on top. “Multiz Alignments of 44 Vertebrates” graphic indicates DNA sequence conservation across 44 species. Distribution of known variants in dbSNP also shown. Graphic taken from the UCSC Genome Browser (genome.ucsc.edu).

Figure 1b. Conservation in the FLCN1 Gene. A 9-species multiple alignment of FLCN1 amino acid sequence. Colored according to ClustalX defaults in the Jalview program (see www.jalview.org for details). ‘Z’ denotes stop codon. There is 72% sequence identity across all 9 species and 89% similarity, indicating very strong conservation.
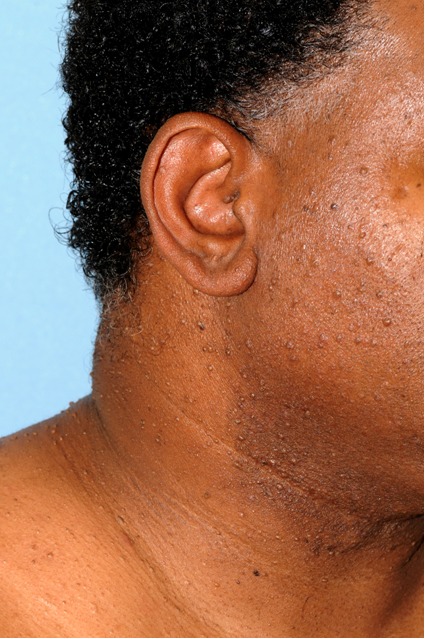
Figure 2. Multiple skin-colored papules on the head and neck representing fibrofolliculomas in a patient with BHD syndrome and Fitzpatrick type V skin.
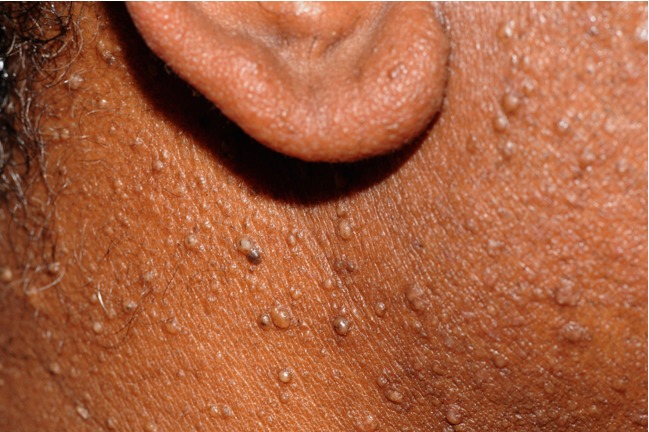
Figure 3. Close-up view of head and neck fibrofolliculomas in a patient with BHD syndrome.

Figure 4. Multiple axillary skin tags (acrochordons) in a patient with BHD syndrome.
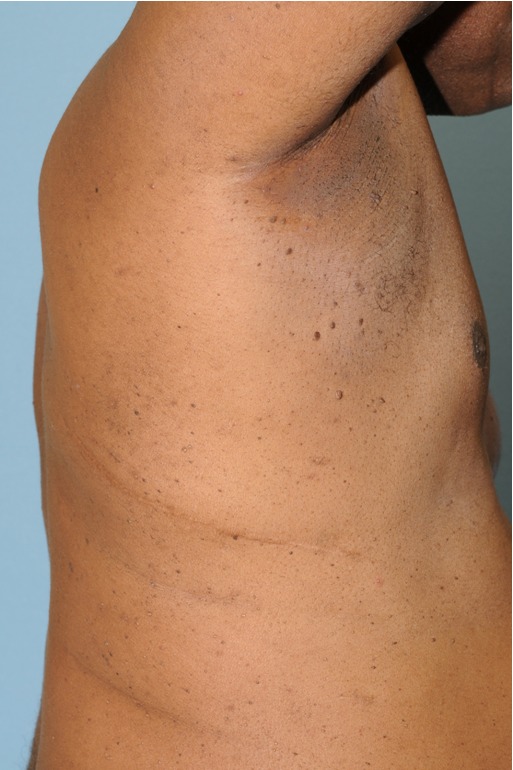
Figure 5. Acrochordons, as seen in this Birt-Hogg-Dubé patient, are commonly seen in patients without the disease as well.
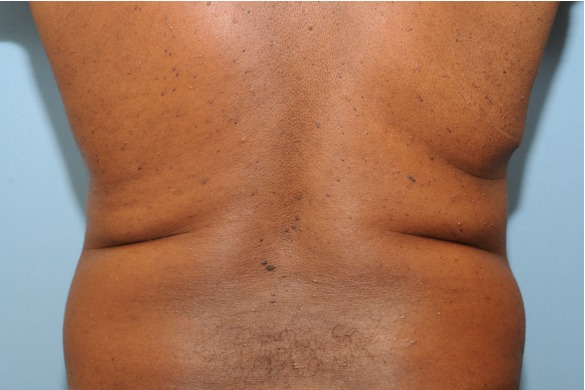
Figure 6. Multiple skin-colored to brown papules on the trunk, also representing fibrofolliculomas in a patient with Birt-Hogg-Dubé. Note that there are fewer lesions than were present on the head and neck.
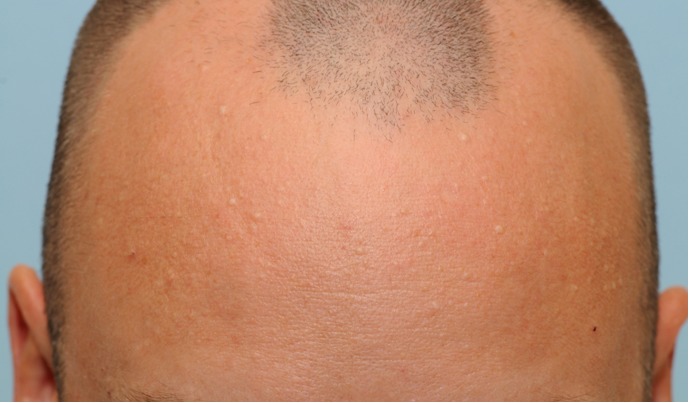
Figure 7. Multiple, waxy, skin-colored to white papules on the forehead in a patient with Birt-Hogg-Dubé syndrome and Fitzpatrick type III skin.
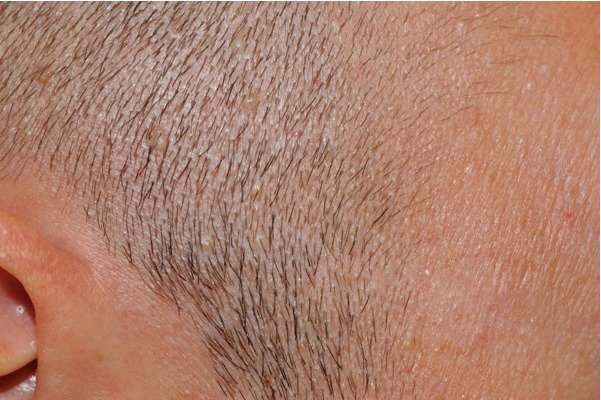
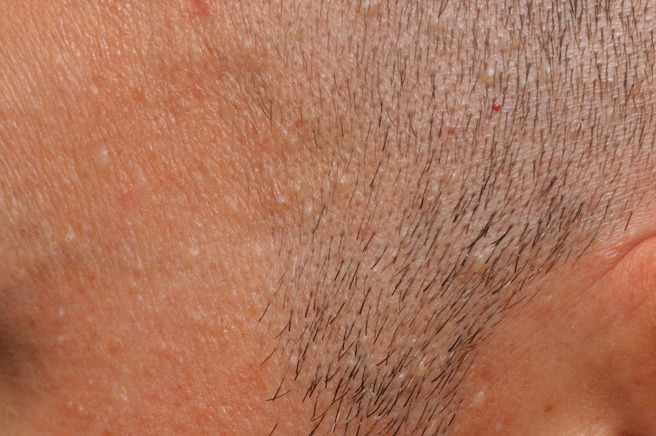
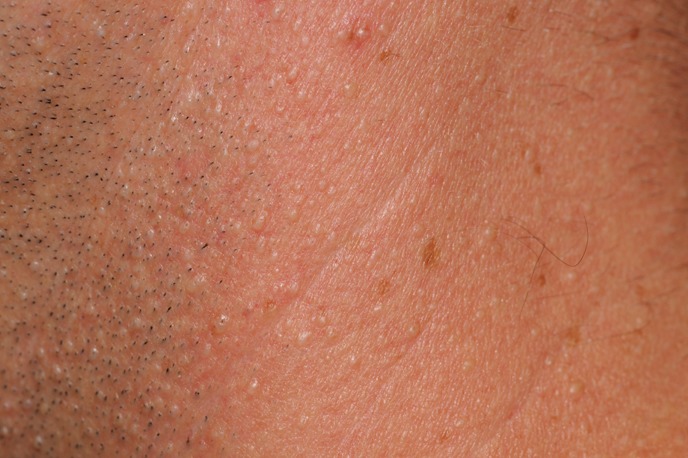
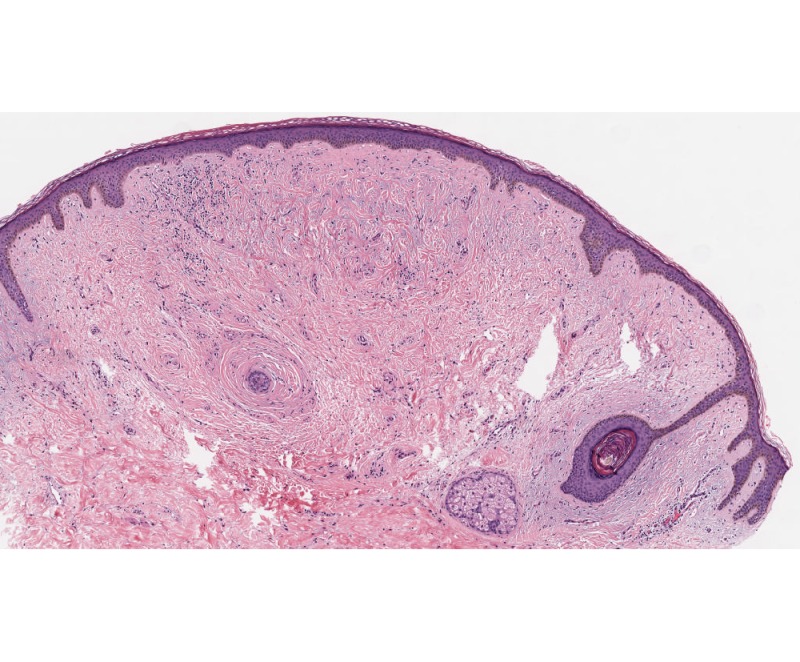
Figure 11. Low-power view showing fibrofolliculoma/trichodiscoma. There is a well-defined dermal nodule comprised of a pauci-cellular fibrous stroma with overlying epidermal effacement in association with a small hair follicle. Note the single anastomosing strand of epithelium extending from a follicular structure on the right. The findings of trichodiscoma predominate in this section.

Figure 12. High-power view showing expansive, fibrous stroma in association with a hair follicle characteristic of trichodiscoma.
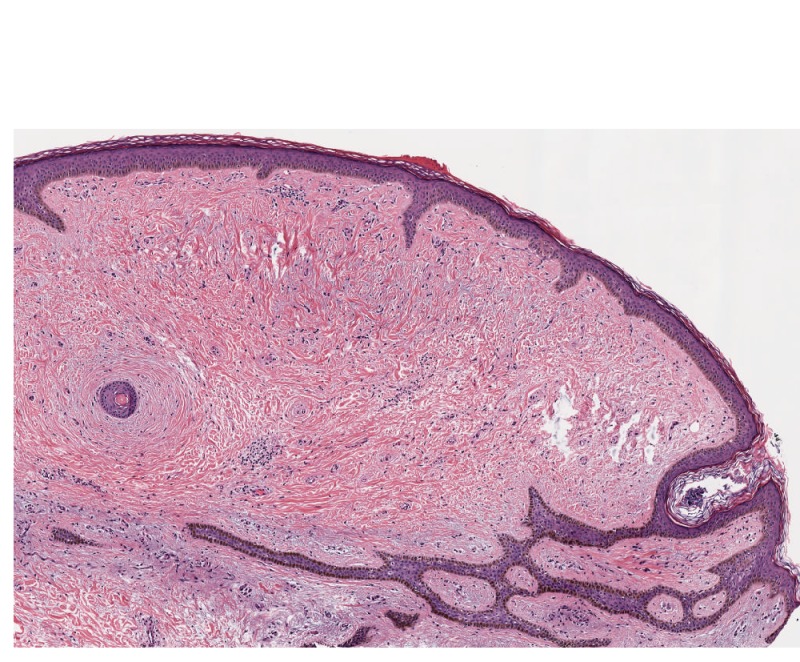
Figure 13. Low-power view showing fibrofolliculoma/trichodiscoma. Subsequent sectioning revealed anastomosing strands of follicular epithelium in association with a plugged hair follicle in a pattern consistent with fibrofolliculoma. In many instances, the central follicular infundibular structure is more dilated and extends deeper in the dermis. Note how these changes chart the more pauci-cellular changes characteristic of trichodiscoma.
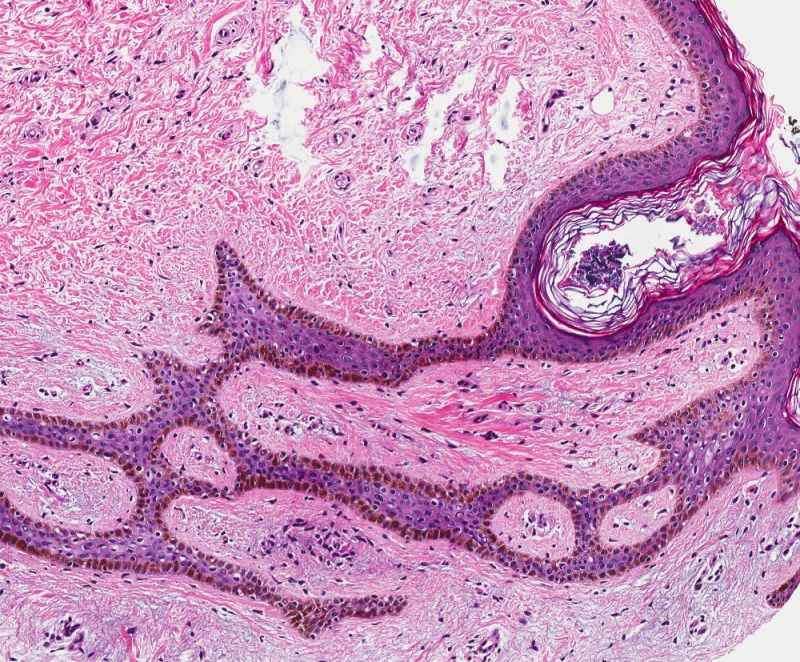
Figure 14. High-power view of fibrofolliculoma/trichodiscoma. Note the anastomosing strands of epithelium in a fibrous stroma characteristic of fibrofolliculoma. Variable sebaceous differentiation can be present.
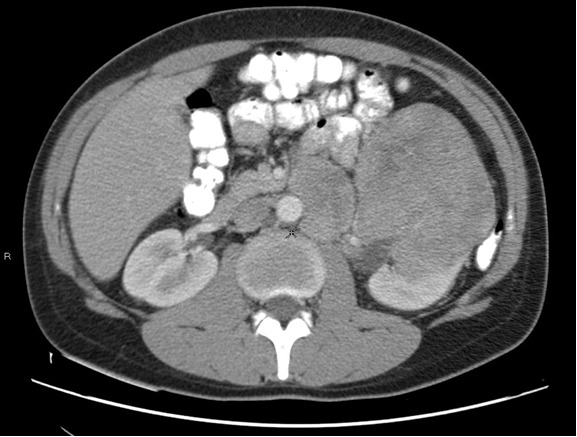
Figure 15. Magnetic resonance imaging showing a tumor of the left kidney in a 27-year-old male with Birt-Hogg-Dubé syndrome. The patient is brother to the patient whose tumor is shown in Figure 16.
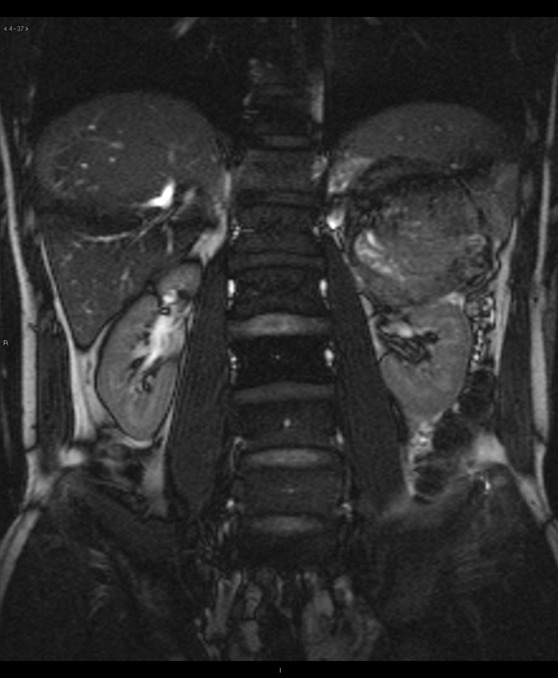
Figure 16. Magnetic resonance imaging showing a large tumor cranial to the left kidney, arising from ectopic renal tissue in the adrenal gland in a 32-year-old male with Birt-Hogg-Dubé syndrome. The patient is the brother of the patient whose tumor is shown in Figure 15.
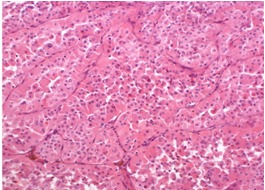
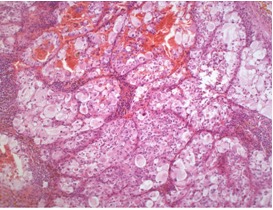
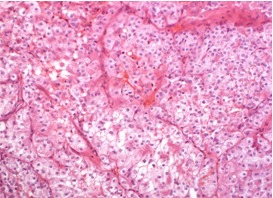
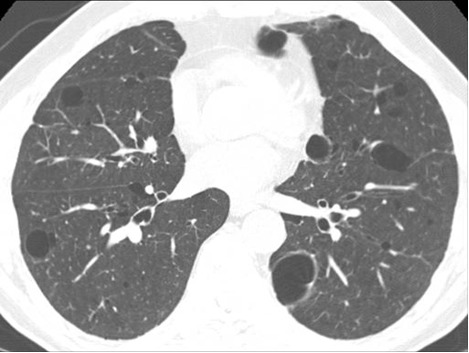
Figure 20. Multiple pulmonary cysts in a patient with Birt-Hogg-Dubé syndrome. See also Figures 21, 22 and Movie Clip 1.
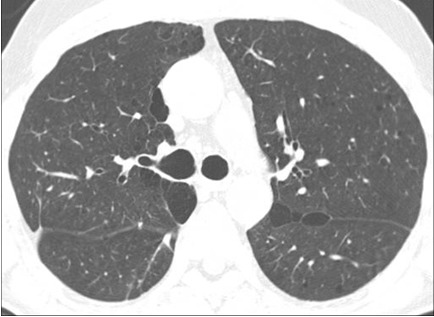
Figure 21. Multiple pulmonary cysts in a patient with Birt-Hogg-Dubé syndrome. See also Figures 20, 22 and Movie Clip 1.
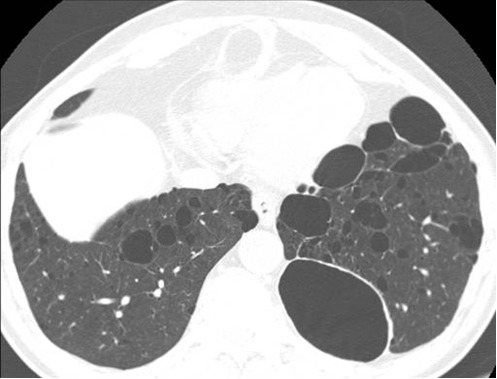
Figure 22. Multiple pulmonary cysts in a patient with Birt-Hogg-Dubé syndrome. See also Figures 20, 21 and Movie Clip 1.
Tables
Table 1. Management considerations in patients with Birt-Hogg-Dubé syndrome.
| Site | Recommendation |
|---|---|
| Kidney | Surveillance beginning at age 20. Interval and technique (computed tomography, ultrasound, or magnetic resonance imaging) to be individualized. |
| Skin | Annual dermatologic examination. |
| Lung | For asymptomatic patients, an initial baseline high resolution chest computated tomography with follow-up study every 3 to 5 years. For patients with symptoms a follow-up should be individualized. In patients undergoing surgery of any kind, the anesthesiologist should be aware of the potential for pneumothoraces. |