It is now well established that the interplay of sequence-specific DNA binding proteins with chromatin components and the subsequent expression of differential genetic programs is the major determinant of developmental decisions. The last years have seen an explosion of basic research that has significantly enhanced our understanding of the basic principles of gene expression control. While many questions are still open, we are now at the stage where we can exploit this knowledge to address questions of how deregulated gene expression and aberrant chromatin programming contributes to disease processes. This article will give a basic introduction into the principles of epigenetics and the determinants of chromatin structure and will discuss the molecular mechanisms of aberrant gene regulation in blood cell diseases, such as inflammation and leukemia.
Introduction
The range of diseases found to have an epigenetic component responsible for aberrant gene regulation is steadily increasing and diseases of blood cells represent some of the best-defined models for studying this type of dysregulation. Many factors control the growth, differentiation and activation status of blood cells and when these are dysregulated the result can be either leukemia with aberrant growth and differentiation, or auto-immune and inflammatory diseases where the immune system is chronically active.
In this chapter we will introduce the basic concepts of chromatin structure and the processes that control gene expression by modifying chromatin. We will draw upon examples from both our own work and the work of others to illustrate the role that chromatin and DNA modifications play in normal gene regulation and in blood cell disease. To introduce some of these concepts we will also discuss the consequences of the reprogramming of the transcriptional regulatory network in leukemic cells that result in abnormal patterns of epigenetic modifications within chromatin.
The Role of Transcription Factors and Chromatin Structure in Establishing Patterns of Gene Expression
Gene expression programs are established during cell differentiation by the concerted actions of sets of transcription factors specific to each cell type and to their state of differentiation and activation. Transcription factors perform multiple functions: they recognize a specific DNA sequence, interact with other transcription factors binding to neighboring DNA-sequences or even several kilobases away,1 respond to extracellular signals and most importantly, recruit nonDNA-binding factor complexes that cooperate to either maintain the active state, or initiate the establishment of an inactive state. These factors, in turn, exert their effects largely at the level of chromatin structure by creating permissive or nonpermissive states. The genome exists naturally in a repressed state by virtue of the fact that regulatory and coding DNA sequences are for the most part occluded by nucleosomes which assemble into highly condensed and inaccessible structures. Before a gene can be expressed, it is necessary to first create accessible sites for the binding of transcription factors required for transcription initiation and secondly, to modify the histones within nucleosomes and reorganize the higher order chromatin structure to create an environment permissive for the passage of RNA-polymerases.
Gene expression programs are typically controlled by transcription factors that are expressed in a temporal sequence during differentiation. Factors such as RUNX1, GATA-2 and PU.1 play pivotal roles in enabling early stages in blood cell differentiation, whereas other factors are responsible for the differentiation of specific hematopoietic lineages. Regulators of differentiation are exemplified by factors such as GATA-3, T-bet and FoxP3 which play important roles in maintaining the balance between effector and regulatory T-cells. Other specific classes of transcription factors only become transcriptional activators as a result of external signals. This is true for inducible factors such as NFAT, AP-1 and NF-κB that play essential roles in mediating responses to immune stimuli. Both the developmentally regulated and the inducible classes of transcription factors can contribute to an aberrantly active immune system.
In addition to exerting transient inducible effects, transcription factors can also introduce stably maintained chromatin alterations. In some cases, transcription factors can establish an imprint within chromatin, creating a memory of a previous stimulatory event which persists after inducible transcription has ceased. For example, we and others have evidence that immune or pro-inflammatory stimuli can induce the formation of modified chromatin structures that can persist many cell cycles after the stimulus is withdrawn and which can remain as long-lived imprints in memory T-cells for example.2,3 Alternatively, specific developmentally regulated transcription factors such as RUNX1 can initiate a cascade of events that become self-perpetuating during blood cell differentiation even after the subsequent removal of the differentiation-initiating factor.4,5
Basic Features of Chromatin Structure
The vast majority of the genome (~99%) exists as nucleosomes comprising ~146 bp of DNA wrapped around an octamer of histone proteins made up of two molecules each of histones H2A, H2B, H3 and H4.6,7 As depicted in Figure 1, nucleosomes assemble in a step-wise manner by first loading two histone H3/H4 dimers as a tetramer onto ~80 bp of DNA and then incorporating two histone H2A/H2B dimers to form a nucleosome particle comprising 146 bp of DNA coiled 1.7 times around the histone octamer core. This model illustrates the fact that nucleosome stability is maintained by multiple contacts along the entire length of the nucleosomal DNA, with each of the eight histone molecules mediating major contacts at two separate sites. It is also significant that this structure has the histone N-terminal tails protruding from the nucleosome core particle, because these tails are the sites of numerous covalent modifications that regulate the structure and function of chromatin.
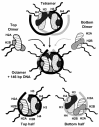
Individual nucleosomes are organized into highly regular arrays where they are separated by linker regions of ~50 bp of DNA, to give an overall average repeat length of ~190-200 bp.8-10 Chains of nucleosomes essentially never exist in a completely decondensed drawn out state, but are arranged in a zig-zag conformation within a highly complex higher order structure. Despite decades of investigation, the precise details of this structure remain elusive. At the first level of folding, nucleosomes coil into 30 nm diameter chromatin fibers (Fig. 2A),8-11 which then assemble further as even more compact structures,12 in which much of the DNA is inaccessible (Fig. 2). This higher order folding is mediated in part by histone H1 which occupies about 20 bp of the linker region between nucleosomes and in part by the positively charged histone tails that extend out from the nucleosome and most likely wrap around the DNA.6

Only about 1% of the genome exists in a decondensed accessible state in any one cell. These accessible regions exist as DNase I hypersensitive sites (DHSs)13 where regulatory factor complexes have opened up localized regions of the chromatin fiber and in most cases this probably involves displacement or disruption of nucleosomes that would otherwise occupy these regions (Fig. 2B). It is also taken for granted that a passing polymerase must transiently create open regions of chromatin (Fig. 2C). This appears to be driven by chromatin remodeling factors and histone-acetyltransferases (HATs) associated with the polymerase complex and is reversed by factors such as histone deacetylases (HDACs) recruited to chromatin that has just been transcribed (Fig. 2C).14,15 One of the purposes of this cycle is to maintain transcribed genes in a predominantly condensed state so as to suppress cryptic promoters.14
The histone tails are subject to a bewildering number of posttranslational modifications. These include lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, lysine ubiquitination, poly-ADP ribosylation, lysine sumoylation, arginine deimination and proline isomerisation.16 Each modification is installed by different families of enzymatic activities, such as HATs, histone methyltransferases (HMTs), kinases or ubiquitin ligases. Depending on the transcriptional state, these modifications are removed by opposing enzymatic activities, such as HDACs and histone demethylases which, like the "writers" of histone modifications, belong to extended families of enzymes with different substrate specificity. Each histone modification serves a distinctive purpose to support either the maintenance of the active (e.g., histone H3 K9 acetylation and K4 methylation) or the inactive (e.g., histone H3 K9 methylation) transcriptional state.
In addition to creating binding sites for various cofactors, the modification of histones also has a direct effect on overall chromatin structure. For example, histone acetylation leads to the neutralization of the positively charged lysines in the histone tails and to a reduced level of compaction, as seen after acetylation of histone H4 K16 (Fig. 2).17 Activation of enhancers and promoters, and the process of transcription, are also accompanied by the replacement of canonical histones with variant histones, such as H2AZ or H3.3 which interact with reduced affinity and thus form less stable nucleosomes This facilitates the displacement of nucleosomes at promoters by the basal transcription machinery.18
The Role of Epigenetic Mechanisms in Cell Differentiation
Research investigating the basis of cell differentiation in the hematopoietic system was instrumental in the development of the concept that stem cells and multi-potent precursor cells activate extended sets of genes at low level prior to differentiation.19 In this context it is important to note that lineage specification and the restriction of developmental potential involve not only the up-regulation of genes important for the development of specific blood cell lineages, but that it is just as important to selectively silence lineage inappropriate genes that exist in an activated state in stem cells. This is a general principle of pattern formation that is common to all multi-cellular organisms and it implies that a regulatory machinery exists which maintains genes in their respective active and silent states and thus maintains cellular identity. It also follows that during the proliferative phases of cell differentiation, such "epigenetic" states have to be faithfully copied during cell division.20 Another important principle of epigenetics is that such regulatory states can be maintained in the absence of the original initiator.21 In the last years, significant progress has been made to identify and characterize the components of the epigenetic regulatory machinery. It is beyond the scope of this article to review its full complexity but the next chapters will review the general principles and discuss the role of the main players: DNA-methylation and histone modifications.
DNA-Methylation
DNA methylation represents one of the most stable epigenetic modifications and plays a major role during development in maintaining specific patterns of gene expression.22-25 In mammals, DNA methylation most commonly involves a symmetrical conversion to 5-methylcytosine on both DNA strands at CG sequences. This modification is introduced by DNA-methyltransferases (DNMTs) that include DNMT3a and DNMT3b which methylate cytosines de novo and DNMT1 which requires a methylated cytosine at one strand of newly replicated DNA and functions to maintain previously installed methylation states during replication.22,26 DNMT3a contacts chromatin in cooperation with DNMT3L and the activating histone H3 K4 methylation modification blocks binding of DNMT3L, and thereby suppresses DNA methylation in active regions.25,27,28
Most of the CG elements in the genome are, by default, maintained in the methylated state if they exist outside of active regions. A side effect of DNA methylation is that CG sequences are relatively rare in the genome, due to the propensity of 5-methylcytosine to mutate to thymidine. The genome also includes many promoter regions that are highly enriched in CG sequences, termed CG islands, that are resistant to DNA methylation. However, these CG islands are themselves significant targets for dysregulation in blood cell diseases and cancer.29 For example, it is common to find aberrant DNA methylation within the CG island promoters of tumor suppressor genes in myelodysplastic disorders,30 which are now sometimes treated with the "epigenetic" drug 5-Azacytidine to reduce genome-wide levels of DNA methylation.31
A recent unbiased genome-wide analysis of sequence patterns characterizing DNA-methylation states identified specific transcription factor motifs at those CG islands that were resistant to de novo methylation. Furthermore, this resistance correlated with the fact that these sites were indeed occupied by these specific transcription factors,32 strongly suggesting that the same factors are responsible for the protection of these sequences from DNA methylation (Fig. 3). How methyl groups are removed from DNA during normal mammalian cell differentiation is still not completely understood and various mechanisms have been proposed.33
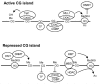
It was recently shown that the balance between the methylated and nonmethylated DNA state at CG islands is controlled in part by proteins containing CXXC motifs that bind to nonmethylated CG sequences (Fig. 3). CXXC domain proteins include the H3K4 HMT Set1 and Cfp1 (also known as CGBP or CXXC1) which associates with the H3K4 HMTs Set1 and MLL1.34-37 CXXC proteins also include Tet1 which a member of the Tet family proteins that hydroxylate 5-methylcytosine.38,39
As modeled in Figure 3, the CXXC and HMT proteins provide potential mechanisms for maintaining regions of high CG content in an unmethylated state. After Tet1 becomes recruited to nonmethylated CG sequences it can presumably modify and eliminate any adjacent methylation of CG sequences. Either MLL1 or the Cfp1/Set1 complex can also bind to nonmethylated CG sequences34,40 and introduce the H3K4 trimethylation mark (Me3H3K4) which suppresses recruitment of the DNMT3a/DNMT3L complex.25 The Me3H3K4 mark is normally associated specifically with transcribed regions,37 but CXXC domains provide a mechanism to introduce this modification even in the absence of transcription. Cfp1 was recently shown to play a major genome-wide role in maintaining CG islands in an active state, being required to introduce the Me3H3K4 modification at CG islands, and it was demonstrated that introduction of an artificial CG island was sufficient to recruit Cfp1 and establish an active Me3H3K4 domain.34
Conversely, CG islands can also be repressed and maintained in a repressed state if they undergo DNA methylation and H3K9 methylation (Fig. 3). These modifications cooperate to maintain the repressed state by promoting recruitment of the H3K9 HMT G9a and DNMTs.23-25 Once methylated, CG islands bind proteins containing methylated DNA-binding domains (MBDs) which recruit H3K9 HMTs and HDACs. However, mechanisms controlling the balance of methylation are highly complex with the same proteins in some cases involved in both activation and repression. Hence, Cfp1 can also recruit DNMT1 and loss of Cfp1 leads to a decrease, not an increase, in levels of DNA methylation within both repeat elements and single copy genes.41 This may indicate that the net balance of Cfp1 function is different at CG islands, where it is required to introduce Me3H3K4, as opposed to interspersed CG elements where it may promote DNA methylation. MBD1 and DNMT1 also each have both CXXC domains and methylated CG binding domains meaning that they can drive repression of CG islands if there is an absence of activating factors.40
Tet family proteins are also targets for mutations in blood cell diseases42 and this could account for the aberrant methylation of CG island promoters in myeloid malignancies.30 The Tet2 gene is frequently mutated in Chronic Myelomonocytic Leukemia (CMML)43,44 and the Tet1 gene (previously termed LCX) is involved in chromosomal translocations in Acute Myeloid Leukemia (AML).45 Interestingly, Tet2 lacks a CXXC domain, meaning that another class of factor is required to direct Tet2 to CG elements and this represents another potential point for epigenetic dysregulation.
Histone Modifications Marking the Inactive Transcriptional State
In the absence of transcriptional activators, the chromatin of genes adopts a heritable silent state by default, or even a heterochromatic state with distinct biochemical features. Heterochromatic genes are highly DNA-methylated and compacted, and harbor inactive histone marks such as methylated histone H3 K9 or K27 which are deposited by the HMTs Su(var)3.9 and EZH2 respectively, the latter being a component of the polycomb family of epigenetic regulators. An important principle is that these DNA and histone modifications serve as binding sites for "readers" of the epigenetic code.46 The best-characterized examples for such interactions are the recognition of methylated CGs by MBD proteins47 and the binding of heterochromatin protein 1 (HP1) family members to trimethylated histone H3 lysine 9.48,49 All of these proteins associate with highly cooperative macromolecular complexes that include histone and DNA-modifying enzymes which either re-install the inactive mark, or remove active marks and thus sustain an inactive chromatin structure. This is exemplified by the findings that (i) DNMT1 and the methyl-binding protein MeCP2 both associate with histone deacetylases (HDACs), (ii) DNMT1 interacts with the H3 K9 HMT G9a50 and (iii) HP1 interacts with both HDACs and DNMTs.51,52
Similar to DNA-methylation, the aberrant deposition of inactive histone marks is a hallmark of disease processes. Since these modifications are normally highly dynamic and get rewritten depending on the transcription cycle and the presence and absence of extracellular signals,53 the deposition of an inactive histone mark per se does not lead to permanent gene silencing. However, as described above, one of the hallmarks of cancer cells is the permanent silencing of CG island promoters of important tumour suppressor genes by aberrant DNA-methylation. It has recently been shown that the binding of such sequences by polycomb complexes and the concomitant deposition of methylated H3 K27 predisposes associated genes to DNA-methylation where they are finally inactivated,54,55 again indicating that the gene silencing machinery operates in a highly cooperative fashion. These experiments also demonstrate that DNA-methylation is the final modification that locks genes into a permanently inactive state.22
Chromatin Modifications Accompanying Gene Activation
Recent genome-wide analyses confirmed that histone modifications are deposited in a combinatorial fashion at the cis-regulatory elements of specific genes, reflecting their differential activities.56,57 The fact that such patterns are not random, and change in response to extracellular stimuli,58 already hints at the fact that the epigenetic regulatory machinery is directed to specific DNA-sequences by sequence specific DNA binding proteins, and their cell-type specific combinatorial action directs the differential deposition of histone modifications.
The architecture of the chromatin fiber is modified in many fundamental ways during the process of gene activation. In the first instance, the regulatory machinery gains entry to the ~1% of the genome that comprises the regulatory elements active in any one cell type by creating highly accessible nucleosome-free regions that exist as DHSs.13 In most cases this involves the cooperative action of different transcription factors, but in some cases the creation of these DHSs is initiated by specialized pioneer factors that have the intrinsic ability to bind to chromatin compacted by histone H1.59 Other specific factors, such as the transcription factors NFAT and NF-κB, are intimately associated with the induction of DHSs within promoter and enhancer elements in response to activation by immune and pro-inflammatory stimuli.60,61 NFAT is a key mediator of T-cell receptor (TCR) signaling, whereas NF-κB is a key mediator of pro-inflammatory signals such as bacterial lipopolysaccharide (LPS). These types of factors play a key role in creating access for and assisting the recruitment of other factors and represent a pivotal point at which the normal tight control over gene expression can be over-ridden in a disease context.
Once bound to DNA, transcription factors recruit a host of chromatin modifying activities. Besides the histone modification machinery described above, they also recruit ATP-dependent nucleosome remodeling complexes such as SWI/SNF and ISWI that either disrupt or reposition nucleosomes and mobilize arrays of nucleosomes.62-65 Remodelers can serve both to create nucleosome-free sites for regulatory factors and polymerases and to render nucleosome organization highly dynamic. Remodeling activities play essential roles in mediating inducible responses within the immune system and we have observed that NFAT-dependent enhancers function in part by mediating long-range mobilization of nucleosomes, creating a highly disorganized and dynamic nucleosome array.66,67 In addition, we showed that transcription factors induced by inflammatory stimuli can activate promoters driving the expression of noncoding RNAs which alter the nucleosomal architecture of cis-regulatory elements by the process of transcription itself.68 Many inducible transcription factors, including for example AP-1 and CREB, which mediate responses within the immune system, have the ability to recruit HATs such as CBP and p300. This typically leads to the creation of a hyperacetylated and more open state at promoters and enhancers.62-65
Similar to mechanisms maintaining the inactive state, histone modifications characteristic for active genes re-enforce the active state by providing interaction modules for the transcription machinery. For example, HATs such as GCN5 and subunits of chromatin remodelers such as Brg1 possess bromodomains that recognize H3 K9 acetylation.69,70 Similarly, H3 K4 tri-methylation is recognized by PHD-finger domains as exemplified by that of TFIID which is part of the basal transcription machinery.71 Moreover, it has recently been shown that this histone modification is required for the maintenance of an active transcriptional state during cell division.72 However, for normal development it is of vital importance that complexes re-enforcing the activated state are tightly regulated. One of the major causes of leukemia is the generation of aberrant epigenetic regulatory proteins as a result of chromosomal translocations. Fusing heterologous domains can lead to the aberrant targeting of activating complexes, as exemplified by a recent study investigating the fusion of the PHD finger of the histone demethylase JARID1A and Nup98.73 The expression of such a dominant-negative fusion protein leads to a targeting of a nonfunctional complex to H3 lysine di/trimethylated sites where it blocks the demethylation of histones. This causes the maintenance of the active state and eventually, a block in cell differentiation and leukemia.
Epigenetics Meets Chronic Inflammation in Leukemia
One of the hallmarks of many cancers is their aberrant growth, which is based on the fact that many tightly regulated growth-controlling signaling processes are dysregulated and constitutively active in these cells. This is achieved by either autocrine/paracrine stimulation of growth factor receptors or the mutation of other molecules involved in transmitting such signals into the nucleus. In blood cells, this involves signaling molecules such as cytokines, cytokine receptors and kinases, as well as transcription factors integrating immune responses. For example, the direct activation of Ras pathways and/or the suppression or mutation of negative regulators of cytokine signaling pathways can lead to activation of genes such as GM-CSF and hypersensitivity to GM-CSF in myeloid malignancies.74-78
The consequence of the chronic activation of inflammatory signals is that transcription factors linking such signals to gene expression control are constitutively active, with the most important factor being NF-κB.79 In human Hodgkin's lymphoma (HL), the constitutive activation of this transcription factor is required for the survival of leukemic cells.80 In the majority of cases, HL cells originate from germinal center B cells, but have lost much of their B cell specific gene expression program.81,82 Interestingly, these cells also express lineage inappropriate genes, including the receptor for colony-stimulating-factor 1 (CSF1R or c-FMS) which is the main growth factor receptor for the macrophage lineage.82,83 Moreover, it was recently shown that these cells also express CSF-1 itself and this autocrine/paracrine stimulation is required for HL cell survival.84 However, the most intriguing result from the same study was that aberrant expression of the CSF1R gene was not driven by its normal promoter, but originated from an aberrantly activated long terminal repeat (LTR) promoter of the THE1B family of repeats located 6.5 kb upstream of the normal transcription start site. LTRs are remnants of retroviral insertions that have remained in the germline. These elements are normally efficiently epigenetically silenced during embryonic development and this silencing is strictly maintained by DNA-methylation and the action of corepressors recruiting HDACs that maintain the presence of inactive histone marks. Moreover, the activation of THE1B elements in HL cells was not restricted to one genomic location, but was a wide-spread phenomenon. As it turned out, HL cells have lost expression of the corepressor MTG8/CBFA2T3 (otherwise known as ETO2). In addition, THE1B elements contain functional binding sites for inducible transcription factors, including NF-κB, which are required to activate LTR-driven promoter activity. The artificial recreation of this situation in non HL cells is sufficient to activate LTR-driven expression, indicating that the loss of epigenetic control combined with constitutive activation of otherwise inducible transcription factors is sufficient to override the safeguards that normally protect cells from the activation of LTR promoters. The consequences of these events are cells of B cell origin that have hijacked myeloid-specific survival signals.
The Role of Epigenetic Mechanisms in Autoimmunity
The role of this article has up until now been to introduce basic concepts of chromatin structure and the epigenetic mechanisms that control the function of the genome in normal cells and in disease. However, we also need to at least touch on the role of epigenetics in autoimmunity, as this is the main theme of this volume, and many specific examples will be discussed in the following chapters. There is now abundant evidence that disturbance of epigenetic mechanisms in the immune system can lead to auto-immune disease, with perhaps the best example being Systemic Lupus Erythematosus (SLE),85-87 which will be discussed in depth in this volume in Chapter 9. This is a disease where there is a prevalence of global DNA hypomethylation and demethylation of the regulatory elements of pro-inflammatory genes such as IL-4 and IL-6 in T-cells.85,88 Furthermore, DNA demethylating agents are able to induce lupus-like symptoms.86 Abnormal patterns of histone acetylation are also found in T-cells from lupus patients. Atopy is another condition where DNA demethylation of genes such as interferon gamma can contribute to autoimmunity.85
Conclusion
The few examples described in this article graphically demonstrate that the interplay of transcription factors with the epigenetic regulatory machinery is at the heart of many disease processes. They also demonstrate that the defects of this interplay are specific for each individual disease. The major challenge for the future will be to delineate the mechanisms common to aberrant gene regulation involved in individual disease processes and identify targets for their correction. A tall order.
Acknowledgements
Our laboratories are supported by Leukaemia and Lymphoma Research, Cancer Research UK, Yorkshire Cancer Research and the BBSRC.
References
- 1.
- de Laat W, Klous P, Kooren J, et al. Three-dimensional organization of gene expression in erythroid cells. Curr Top Dev Biol. 2008;82:117–139. [PubMed: 18282519]
- 2.
- Gialitakis M, Arampatzi P, Makatounakis T, et al. Mol Cell Biol. 2010. Interferon gamma dependent transcriptional memory via relocalization of a gene locus to PML nuclear bodies. [PMC free article: PMC2849471] [PubMed: 20123968]
- 3.
- Mirabella F, Baxter EW, Boissinot M, et al. The Human IL-3/Granulocyte-Macrophage Colony-Stimulating Factor Locus Is Epigenetically Silent in Immature Thymocytes and Is Progressively Activated during T-Cell Development. J Immunol. 2010;184(6):3043–3054. [PubMed: 20147630]
- 4.
- Chen MJ, Yokomizo T, Zeigler BM, et al. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457(7231):887–891. [PMC free article: PMC2744041] [PubMed: 19129762]
- 5.
- Hoogenkamp M, Lichtinger M, Krysinska H, et al. Early chromatin unfolding by RUNX1: a molecular explanation for differential requirements during specification versus maintenance of the hematopoietic gene expression program. Blood. 2009;114(2):299–309. [PMC free article: PMC2714206] [PubMed: 19339695]
- 6.
- Zlatanova J, Leuba SH, van Holde K. Chromatin structure revisited. Crit Rev Eukaryot Gene Expr. 1999;9(3-4):245–255. [PubMed: 10651241]
- 7.
- Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. [PubMed: 9305837]
- 8.
- Robinson PJ, Fairall L, Huynh VA, et al. EM measurements define the dimensions of the "30-nm" chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci USA. 2006;103(17):6506–6511. [PMC free article: PMC1436021] [PubMed: 16617109]
- 9.
- Schalch T, Duda S, Sargent DF, et al. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436(7047):138–141. [PubMed: 16001076]
- 10.
- Williams SP, Athey BD, Muglia LJ, et al. Chromatin fibers are left-handed double helices with diameter and mass per unit length that depend on linker length. Biophys J. 1986;49(1):233–248. [PMC free article: PMC1329627] [PubMed: 3955173]
- 11.
- Woodcock CL, Frado LL, Rattner JB. The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J Cell Biol. 1984;99(1 Pt 1):42–52. [PMC free article: PMC2275637] [PubMed: 6736132]
- 12.
- Hu Y, Kireev I, Plutz M, et al. Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J Cell Biol. 2009;185(1):87–100. [PMC free article: PMC2700507] [PubMed: 19349581]
- 13.
- Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. [PubMed: 3052270]
- 14.
- Carrozza MJ, Li B, Florens L, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123(4):581–592. [PubMed: 16286007]
- 15.
- Zippo A, Serafini R, Rocchigiani M, et al. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138(6):1122–1136. [PubMed: 19766566]
- 16.
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. [PubMed: 17320507]
- 17.
- Shogren-Knaak M, Ishii H, Sun JM, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. [PubMed: 16469925]
- 18.
- Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21(12):1519–1529. [PMC free article: PMC1891429] [PubMed: 17575053]
- 19.
- Enver T, Greaves M. Loops, lineage and leukemia. Cell. 1998;94(1):9–12. [PubMed: 9674421]
- 20.
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10(3):192–206. [PubMed: 19234478]
- 21.
- Berger SL, Kouzarides T, Shiekhattar R, et al. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. [PMC free article: PMC3959995] [PubMed: 19339683]
- 22.
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. [PubMed: 11782440]
- 23.
- Berger J, Bird A. Role of MBD2 in gene regulation and tumorigenesis. Biochem Soc Trans. 2005;33(Pt 6):1537–1540. [PubMed: 16246164]
- 24.
- Bird A. The methyl-CpG-binding protein MeCP2 and neurological disease. Biochem Soc Trans. 2008;36(Pt 4):575–583. [PubMed: 18631120]
- 25.
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10(5):295–304. [PubMed: 19308066]
- 26.
- Chen T, Li E. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. [PubMed: 16570848]
- 27.
- Ooi SK, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–717. [PMC free article: PMC2650820] [PubMed: 17687327]
- 28.
- Jia D, Jurkowska RZ, Zhang X, et al. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449(7159):248–251. [PMC free article: PMC2712830] [PubMed: 17713477]
- 29.
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. [PMC free article: PMC2802667] [PubMed: 19752007]
- 30.
- Boultwood J, Wainscoat JS. Gene silencing by DNA methylation in haematological malignancies. Br J Haematol. 2007;138(1):3–11. [PubMed: 17489980]
- 31.
- Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem. 2009;16(17):2075–2085. [PubMed: 19519382]
- 32.
- Gebhard C, Benner C, Ehrich M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res. 70(4):1398–1407. [PubMed: 20145141]
- 33.
- Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166. [PMC free article: PMC3137514] [PubMed: 19659441]
- 34.
- Thomson JP, Skene PJ, Selfridge J, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464(7291):1082–1086. [PMC free article: PMC3730110] [PubMed: 20393567]
- 35.
- Ansari KI, Mishra BP, Mandal SS. Human CpG binding protein interacts with MLL1, MLL2 and hSet1 and regulates Hox gene expression. Biochim Biophys Acta. 2008;1779(1):66–73. [PubMed: 18082152]
- 36.
- Tate CM, Lee JH, Skalnik DG. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J. 2010;277(1):210–223. [PMC free article: PMC2806598] [PubMed: 19951360]
- 37.
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20(3):341–348. [PMC free article: PMC2504688] [PubMed: 18508253]
- 38.
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. [PMC free article: PMC2715015] [PubMed: 19372391]
- 39.
- Loenarz C, Schofield CJ. Oxygenase catalyzed 5-methylcytosine hydroxylation. Chem Biol. 2009;16(6):580–583. [PubMed: 19549596]
- 40.
- Allen MD, Grummitt CG, Hilcenko C, et al. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 2006;25(19):4503–4512. [PMC free article: PMC1589984] [PubMed: 16990798]
- 41.
- Carlone DL, Lee JH, Young SR, et al. Reduced genomic cytosine methylation and defective cellular differentiation in embryonic stem cells lacking CpG binding protein. Mol Cell Biol. 2005;25(12):4881–4891. [PMC free article: PMC1140577] [PubMed: 15923607]
- 42.
- Mullighan CG. TET2 mutations in myelodysplasia and myeloid malignancies. Nat Genet. 2009;41(7):766–767. [PubMed: 19557078]
- 43.
- Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2 and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. [PMC free article: PMC2710942] [PubMed: 19420352]
- 44.
- Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–6410. [PMC free article: PMC2710933] [PubMed: 19372255]
- 45.
- Ono R, Taki T, Taketani T, et al. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002;62(14):4075–4080. [PubMed: 12124344]
- 46.
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. [PubMed: 11498575]
- 47.
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. [PubMed: 16403636]
- 48.
- Lachner M, OCarroll D, Rea S, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. [PubMed: 11242053]
- 49.
- Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–124. [PubMed: 11242054]
- 50.
- Esteve PO, Chin HG, Smallwood A, et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20(22):3089–3103. [PMC free article: PMC1635145] [PubMed: 17085482]
- 51.
- Smallwood A, Esteve PO, Pradhan S, et al. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21(10):1169–1178. [PMC free article: PMC1865489] [PubMed: 17470536]
- 52.
- Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. [PubMed: 9620804]
- 53.
- Metivier R, Penot G, Hubner MR, et al. Estrogen receptor-alpha directs ordered, cyclical and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115(6):751–763. [PubMed: 14675539]
- 54.
- Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 premarks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–236. [PubMed: 17200670]
- 55.
- Ohm JE, McGarvey KM, Yu X, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39(2):237–242. [PMC free article: PMC2744394] [PubMed: 17211412]
- 56.
- Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40(7):897–903. [PMC free article: PMC2769248] [PubMed: 18552846]
- 57.
- Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–1031. [PMC free article: PMC2750862] [PubMed: 19698979]
- 58.
- Ghisletti S, Barozzi I, Mietton F, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. [PubMed: 20206554]
- 59.
- Cirillo LA, Lin FR, Cuesta I, et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell. 2002;9(2):279–289. [PubMed: 11864602]
- 60.
- Cockerill PN. NFAT is well placed to direct both enhancer looping and domain-wide models of enhancer function. Sci Signal. 2008;1(13) [PubMed: 18385038]
- 61.
- Hogan PG, Chen L, Nardone J, et al. Transcriptional regulation by calcium, calcineurin and NFAT. Genes Dev. 2003;17(18):2205–2232. [PubMed: 12975316]
- 62.
- Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006;22(6):320–329. [PubMed: 16631276]
- 63.
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. [PubMed: 17362198]
- 64.
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. [PubMed: 17522673]
- 65.
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. [PubMed: 17320508]
- 66.
- Johnson BV, Bert AG, Ryan GR, et al. GM-CSF enhancer activation requires cooperation between NFAT and AP-1 elements and is associated with extensive nucleosome reorganization. Mol Cell Biol. 2004;24(18):7914–7930. [PMC free article: PMC515070] [PubMed: 15340054]
- 67.
- Bert AG, Johnson BV, Baxter EW, et al. A modular enhancer is differentially regulated by GATA and NFAT elements that direct different tissue-specific patterns of nucleosome positioning and inducible chromatin remodeling. Mol Cell Biol. 2007;27(8):2870–2885. [PMC free article: PMC1899937] [PubMed: 17283044]
- 68.
- Lefevre P, Witham J, Lacroix CE, et al. The LPS-induced transcriptional upregulation of the chicken lysozyme locus involves CTCF eviction and noncoding RNA transcription. Mol Cell. 2008;32(1):129–139. [PMC free article: PMC2581490] [PubMed: 18851839]
- 69.
- Hassan AH, Prochasson P, Neely KE, et al. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111(3):369–379. [PubMed: 12419247]
- 70.
- Marmorstein R, Berger SL. Structure and function of bromodomains in chromatin-regulating complexes. Gene. 2001;272(1-2):1–9. [PubMed: 11470504]
- 71.
- Vermeulen M, Mulder KW, Denissov S, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131(1):58–69. [PubMed: 17884155]
- 72.
- Muramoto T, Muller I, Thomas G, et al. Methylation of H3K4 Is Required for Inheritance of Active Transcriptional States. Curr Biol. 20(5):397–406. [PubMed: 20188556]
- 73.
- Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459(7248):847–851. [PMC free article: PMC2697266] [PubMed: 19430464]
- 74.
- Bowen DT. Chronic myelomonocytic leukemia: lost in classification? Hematol Oncol. 2005;23(1):26–33. [PubMed: 16149105]
- 75.
- Russell NH. Autocrine growth factors and leukaemic haemopoiesis. Blood Rev. 1992;6(3):149–156. [PubMed: 1422283]
- 76.
- Birnbaum RA, OMarcaigh A, Wardak Z, et al. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol Cell. 2000;5(1):189–195. [PubMed: 10678181]
- 77.
- Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108(7):2349–2357. [PMC free article: PMC1895567] [PubMed: 16763213]
- 78.
- Ramshaw HS, Bardy PG, Lee MA, et al. Chronic myelomonocytic leukemia requires granulocyte-macrophage colony-stimulating factor for growth in vitro and in vivo. Exp Hematol. 2002;30(10):1124–1131. [PubMed: 12384142]
- 79.
- Van Waes C. Nuclear factor-kappaB in development, prevention and therapy of cancer. Clin Cancer Res. 2007;13(4):1076–1082. [PubMed: 17317814]
- 80.
- Bargou RC, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkins disease tumor cells. J Clin Invest. 1997;100(12):2961–2969. [PMC free article: PMC508507] [PubMed: 9399941]
- 81.
- Kuppers R. The biology of Hodgkins lymphoma. Nat Rev Cancer. 2009;9(1):15–27. [PubMed: 19078975]
- 82.
- Mathas S, Janz M, Hummel F, et al. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol. 2006;7(2):207–215. [PubMed: 16369535]
- 83.
- Bonifer C, Hume DA. The transcriptional regulation of the Colony-Stimulating Factor 1 Receptor (csf1r) gene during hematopoiesis. Front Biosci. 2008;13:549–560. [PubMed: 17981568]
- 84.
- Lamprecht B, Walter K, Kreher S, et al. Derepression of an endogenous long terminal repeat activates the CSF1R protooncogene in human lymphoma. Nature Medicine. 2010;16(5):571–579. [PubMed: 20436485]
- 85.
- Szyf M. Clin Rev Allergy Immunol. 2009. Epigenetic therapeutics in autoimmune disease. [PubMed: 19644776]
- 86.
- Zhou Y, Lu Q. DNA methylation in T-cells from idiopathic lupus and drug-induced lupus patients. Autoimmun Rev. 2008;7(5):376–383. [PubMed: 18486925]
- 87.
- Richardson B. DNA methylation and autoimmune disease. Clin Immunol. 2003;109(1):72–79. [PubMed: 14585278]
- 88.
- Mi XB, Zeng FQ. Hypomethylation of interleukin-4 and -6 promoters in T-cells from systemic lupus erythematosus patients. Acta Pharmacol Sin. 2008;29(1):105–112. [PubMed: 18158872]
Publication Details
Author Information and Affiliations
Authors
Bonifer Constanze and Peter N Cockerill.
Affiliations
Section of Experimental Haematology, Leeds Institute of Molecular Medicine, University of Leeds, St James's University Hospital, Leeds, UK. Emails: ku.ca.sdeel@refinob.c and; Email: ku.ca.sdeel@llirekcoc.n.p
Notes
Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Constanze B, Cockerill PN. Chromatin Mechanisms Regulating Gene Expression In Health And Disease. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.


