Summary
Clinical characteristics.
Allan-Herndon-Dudley syndrome (AHDS), an X-linked disorder, is characterized in males by neurologic findings (hypotonia and feeding difficulties in infancy, developmental delay / intellectual disability ranging from mild to profound) and later-onset pyramidal signs, extrapyramidal findings (dystonia, choreoathetosis, paroxysmal movement disorder, hypokinesia, masked facies), and seizures, often with drug resistance. Additional findings can include dysthyroidism (manifest as poor weight gain, reduced muscle mass, and variable cold intolerance, sweating, elevated heart rate, and irritability) and pathognomonic thyroid test results. Most heterozygous females are not clinically affected but may have minor thyroid test abnormalities.
Diagnosis/testing.
The diagnosis of AHDS is established in a male proband with suggestive findings and a hemizygous SLC16A2 pathogenic variant identified by molecular genetic testing, and in a female proband by identification of a heterozygous pathogenic variant in SLC16A2.
Management.
Treatment of manifestations: Multidisciplinary team to provide standard care for hypotonia, poor feeding, DD/ID, spasticity, and extrapyramidal movement disorders. Standard treatment with anti-seizure medication by an experienced neurologist. Thyroid hormone replacement therapy during childhood has no beneficial effect and could be dangerous by worsening dysthyroidism.
Surveillance: In children, assess the following every six months until age four years, then once a year: developmental progress & educational needs; neurologic examination for new manifestations (e.g., seizures, changes in tone, movement disorders); spine for scoliosis and hips for dislocation; mobility and self-help skills.
Agents/circumstances to avoid: Administration of L-T4 or L-T3 alone can exacerbate the high serum T3 levels and the resulting hypermetabolism.
Therapies under investigation: A T3 analog TRIAC (acide 3,3',5-triiodothyroacetique) has been tested for a maximum of one year in an international multicentric study of 46 individuals with AHDS. The main objective, normalization of the free T3 blood level, was achieved. Other favorable findings were increased body weight; decreased heart rate, systolic blood pressure, and hypertension; and improved development in seven children, two of whom had started TRIAC treatment before age four years and achieved independent sitting and full head control after 12 months of treatment.
Genetic counseling.
AHDS is inherited in an X-linked manner. If the mother of a proband has an SLC16A2 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the variant will be heterozygotes (carriers) and usually will not be clinically affected but may have minor thyroid test abnormalities. Once the SLC16A2 pathogenic variant has been identified in an affected family member, carrier testing of at-risk female relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Formal diagnostic criteria for Allan-Herndon-Dudley syndrome have not been established.
Suggestive Findings
Allan-Herndon-Dudley syndrome (AHDS) should be considered in males with the following clinical findings, brain imaging, and thyroid hormone profiles.
Clinical Findings
Neurologic
- Onset before age two years often with hypotonia and feeding difficulties
- Developmental delay / intellectual disability ranging from mild to profound intellectual disability
- Extrapyramidal findings: dystonia, choreoathetosis, paroxysmal movement disorder, hypokinesia, hypomimia (masked facies)
- Pyramidal signs
- Late-onset seizures, often with drug resistance
Dysthyroidism
- Poor weight gain
- Reduced muscle mass
- Variably present: cold intolerance, sweating, elevated heart rate, irritability
Craniofacial. Common facial findings that may be attributed to prenatal and infantile hypotonia include ptosis, open mouth, and a tented upper lip. Ear length is above the 97th centile in about half of adults. Cup-shaped ears, thickening of the nose and ears, upturned earlobes, and a decrease in facial creases and a long face are also reported.
Laboratory Findings
Males with AHDS have pathognomonic thyroid test results (Figure 1) including the following:

Figure 1.
Thyroid profiles of 24 patients with AHDS (black triangles) compared to 25 male patients with other genetically defined intellectual disability (gray circles). Serum levels of: A. TSH *
- High serum 3,3',5-triiodothyronine (usually free T3) concentration and low serum 3,3',5'-triiodothyronine (reverse T3, or rT3) concentrationNote: All males with SLC16A2 pathogenic variants had high serum T3 concentration and, when obtained, low serum rT3 concentration. This holds true for both total and free hormone concentrations in serum.
- Serum tetraiodothyronines (total T4 and free T4) concentration are often reduced, but may be within the low normal range
- Free T3/T4 ratio >0.75 (expressed as mmol/mmol) [Remerand et al 2019]
- Serum TSH concentrations that are normal or slightly elevated (Figure 1) [Refetoff & Dumitrescu 2007, Dumitrescu & Refetoff 2009, Remerand et al 2019]
Imaging
Brain MRI in children under age five years usually shows severely delayed myelination mimicking hypomyelination, which subsequently improves over time (Figure 2) [Holden et al 2005, Kakinuma et al 2005, Sijens et al 2008, Vaurs-Barrière et al 2009, Gika et al 2010, Tsurusaki et al 2011, Tonduti et al 2013, Remerand et al 2019].
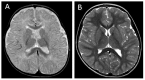
Figure 2.
(A) T2-weighted sequences of the brain MRI of a child age 12 months with AHDS showing diffusely abnormal white matter; (B) same child at age 7 years showing improved myelination with time
Note: Early reports of normal brain MRI findings in this disorder were from older individuals. Cerebral atrophy is also a frequent sign associated with hypomyelination.
Establishing the Diagnosis
Male proband. The diagnosis of AHDS is established in a male proband with suggestive findings and a hemizygous SLC16A2 pathogenic (or likely pathogenic) variant identified by molecular genetic testing (see Table 1).
Female proband. The diagnosis of AHDS is usually established in a female proband by identification of a heterozygous pathogenic (or likely pathogenic) variant in SLC16A2 by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a hemizygous or heterozygous SLC16A2 variant of uncertain significance does not establish or rule out a diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of AHDS is broad, individuals with the distinctive clinical and laboratory findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of AHDS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SLC16A2 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants. If no pathogenic variant is found, gene-targeted deletion/duplication analysis is usually performed next to detect intragenic deletions or duplications.
An intellectual disability, leukodystrophy, or abnormal movement disorder multigene panel that includes SLC16A2 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder, a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Allan-Herndon-Dudley Syndrome
Clinical Characteristics
Clinical Description
Allan-Herndon-Dudley syndrome (AHDS), an X-linked disorder, is characterized in males by neurologic findings (hypotonia and feeding difficulties in infancy, developmental delay [DD] / intellectual disability [ID]) and later-onset pyramidal signs, extrapyramidal findings, and seizures, often with drug resistance. Dysthyroidism can manifest as poor weight gain, reduced muscle mass and variable cold intolerance, sweating, elevated heart rate, irritability, and pathognomonic thyroid test results. Most heterozygous females are not clinically affected but may have minor thyroid test abnormalities.
Affected Males
To date, information on about 200 individuals with a pathogenic variant in SLC16A2 has been published [Groeneweg et al 2019, Remerand et al 2019]. The following description of the phenotypic features associated with this condition is based on the report by Remerand et al [2019].
Table 2.
Select Features of Allan-Herndon-Dudley Syndrome in Affected Males
Prenatal/neonatal findings. Infants with AHDS have normal length, weight, and head circumference at birth. Hypotonia, feeding difficulties and early weight gain deficiency can appear in the first weeks or months of life. Prolonged neonatal jaundice has recently been reported.
Growth. Weight gain lags behind linear growth; low weight is a frequent feature Linear growth is frequently normal initially, but between 10 and 30% of males with time have short stature; microcephaly becomes apparent with age.
Developmental delay / intellectual disability. Most affected males have profound-to-severe intellectual disability with no acquisition of walking; most affected males never speak or may develop only garbled sounds secondary to severely dysarthric speech.
Less frequently, affected males have mild-to-moderate intellectual disability, and develop the ability to walk (with or without aid) and use of language allowing academic learning with aid.
Neuromuscular. Truncal hypotonia, a main feature of AHDS, persists into adulthood. Adults are described with "limber neck" or poor head control.
Progressive hypertonicity of the limbs with brisk reflexes, ankle clonus, and extensor plantar responses (Babinski sign) leads to spastic quadriplegia and joint contractures.
Overall muscle mass (particularly proximally) is reduced and associated with generalized muscle weakness.
It is common for affected males to experience purposeless movements described as dystonic and/or athetoid and characteristic paroxysms or kinesigenic dyskinesias [Brockmann et al 2005, Fuchs et al 2009]. These can be triggered by somatosensory stimuli, including changing clothes or diaper, or lifting the affected child. During attacks, the body extends and the mouth opens; stretching or flexing of the limbs lasts as long as one to two minutes.
Some authors also reported abnormal movements as ataxia [Schwartz et al 2005].
Seizures typically begin during infancy or early childhood. Drug resistance is common [Schwartz & Stevenson 2007, Remerand et al 2019].
Rotary nystagmus and disconjugate eye movements have been reported but are not common [Dumitrescu et al 2004, Remerand et al 2019].
Skeletal. Pectus excavatum and kyphoscoliosis are most likely the result of hypotonia and reduced muscle mass.
Behavior. Generally, affected individuals are attentive, friendly, and docile. They are not aggressive or destructive.
Other. Peripheral dysthyroidism can be expressed as cold intolerance, sweating, intestinal transit disorders, tachycardia, high blood pressure, and sleep disorders.
Life span. Early death has occurred in some individuals, usually caused by recurrent infections and/or aspiration pneumonia. In a few instances survival beyond age 70 years has been reported.
Affected Heterozygous Females
Heterozygous females are generally asymptomatic and have no specific phenotypic findings. About 25% of heterozygous female have an abnormal thyroid profile with elevated T3 levels without any neurologic manifestations [Ramos et al 2011, García-de Teresa et al 2015].
Developmental delay and intellectual disability have been reported in heterozygous females in rare instances, perhaps due to skewed X-chromosome inactivation [Dumitrescu et al 2004, Schwartz et al 2005, Herzovich et al 2007, García-de Teresa et al 2015]. One female had typical features of AHDS with a de novo translocation disrupting SLC16A2 and unfavorable nonrandom X-chromosome inactivation [Frints et al 2008]. One exception of note was the finding in one female of a whole or partial deletion of one X chromosome and a SLC16A2 pathogenic variant on the other X chromosome. However, whether a causative relationship exists between SLC16A2 pathogenic variants and cognitive impairments in heterozygous females has yet to be proven [Schwartz et al 2005].
Genotype-Phenotype Correlations
It has been repeatedly reported that the severity of the clinical phenotype is related to the residual transport capacity of the mutated MCT8 protein. Large deletions in SLC16A2 are assumed to result in complete inactivation of MCT8 and a consequently severe phenotype. While the most frequent large SLC16A2 deletions are of exon 1, deletions of exons 2-4, exons 2-6, exon 3, exons 3-4, and exon 6 have also been reported [Friesema et al 2004, Jansen et al 2007, Vaurs-Barrière et al 2009, Visser et al 2009, Friesema et al 2010, Gika et al 2010, Zung et al 2011, Yamamoto et al 2013, Anık et al 2014, García-de Teresa et al 2015, Remerand et al 2019].
Several SLC16A2 pathogenic missense variants and an in-frame single amino-acid deletion (Table 7) have been associated with considerable residual MCT8 thyroid hormone transport capacity and a milder clinical phenotype, including some speech development, some reading/writing ability, and/or the ability to walk with or without support [Schwartz et al 2005, Jansen et al 2008, Vaurs-Barrière et al 2009, Visser et al 2009, Visser et al 2013, Philips et al 2014, Novara et al 2017, Masnada et al 2019, Remerand et al 2019]. Independent walking and speech development are unusual in affected males with other pathogenic variants.
Nomenclature
This condition was named MCT8-specific thyroid hormone cell-membrane transporter deficiency following identification of the causative gene, SLC16A2, and the defect in thyroid hormone metabolism.
Because of the overlap of clinical findings in individuals with an SLC16A2 pathogenic variant and Allan-Herndon-Dudley syndrome (AHDS), Schwartz et al [2005] analyzed SLC16A2 and identified variants in six families with MCT8-specific thyroid hormone cell-membrane transporter deficiency. Thus, AHDS and MCT8-specific thyroid hormone cell-membrane transporter deficiency are synonyms.
Prevalence
Prevalence of Allan-Herndon-Dudley syndrome (AHDS) is unknown; however, the identification of more than 160 affected individuals in approximately 15 years suggests that the syndrome is more common than previously thought.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in SLC16A2.
Differential Diagnosis
Many disorders demonstrate hypotonia and severe intellectual disability in an X-linked or autosomal recessive inheritance pattern. The main differential diagnoses, described in Table 3, also demonstrate dystonia, spasticity, seizures, or other features that overlap with the neurologic phenotype of Allan-Herndon-Dudley syndrome. More widely, all diseases leading to X-linked intellectual disability, hypomyelinating leukodystrophies or precocious dystonia should be considered as differential diagnoses.
Table 3.
Genes of Interest in the Differential Diagnosis of Allan-Herndon-Dudley Syndrome (AHDS)
Management
No current published guidelines exist to establish the extent of disease or proper management in an individual diagnosed with Allan-Herndon-Dudley syndrome (AHDS). The following recommendations are based on current literature and the authors' experience.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Allan-Herndon-Dudley syndrome (AHDS), the evaluations summarized Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Allan-Herndon-Dudley Syndrome
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Allan-Herndon-Dudley Syndrome (AHDS)
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability (DD/ID) in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically by an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Surveillance
Table 6.
Recommended Surveillance for Individuals with Allan-Herndon-Dudley Syndrome
Agents/Circumstances to Avoid
Administration of L-T4 or L-T3 alone can exacerbate the high serum T3 levels and the resulting hypermetabolism.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Two unaffected heterozygous pregnant women with unaffected fetuses were treated with L-T4 in the second half of pregnancy [Ramos et al 2011]. It is unclear if this had any effect, either beneficial or detrimental, on the fetus. Of note, many unaffected heterozygous mothers have given birth to normal unaffected children without any prenatal treatment.
Therapies Under Investigation
Recently, an T3 analog TRIAC (acide 3,3',5-triiodothyroacetique) has been tested in an international multicentric study, coordinated by the Erasmus University (Rotterdam, Netherlands) [Groeneweg et al 2019]; 46 persons with AHDS were included and treated with a maximum of one year of TRIAC.
The main objective was the normalization of the free T3 blood level; T3 concentration declined significantly (reduction of 61% of baseline). Other findings: a mean increase of body weight of 2.7 kg, a mean decrease of heart rate over 24 hours of five beats per minute, a mean decrease of systolic blood pressure from the 78th centile to the 61st centile, and a mean decrease of hypertension from 34% to 9%.
On neurologic examination, of the seven individuals with a completely inactivating SLC16A2 variant who had started TRIAC treatment before age four years, two reached independent sitting and achieved full head control after 12 months of treatment.
Seven mild and transient adverse effects related to TRIAC occurred in six individuals: three had increased perspiration and three reported irritability.
Beginning in 2017 the European Medicines Agency (EMA) granted TRIAC orphan designation for the treatment of AHDS (EMA/695502/2017).
To follow this first clinical study, an international Phase II trial (NCT02396459) to investigate the effects of TRIAC on neurodevelopmental outcomes in children younger than 30 months with AHDS will begin recruiting in early 2020.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Allan-Herndon-Dudley syndrome (AHDS) is inherited in an X-linked manner.
Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for the SLC16A2 pathogenic variant; therefore, he does not require further evaluation/testing.
- In a study of 24 affected individuals, 17 males had inherited the SLC16A2 pathogenic variant from their mother [Remerand et al 2019].
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected child and no other affected relatives and if the SLC16A2 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier) or the affected male may have a de novo SLC16A2 pathogenic variant, in which case the mother is not a carrier. De novo variants have been reported in AHDS [Dumitrescu et al 2004; Author, personal communication]. In a study of 24 affected individuals, seven had a de novo SLC16A2 pathogenic variant [Remerand et al 2019].
Sibs of a male proband. The risk to the sibs depends on the genetic status of the mother:
- If the mother of the proband has an SLC16A2 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the variant will be carriers and will usually not be clinically affected but may have minor thyroid test abnormalities (see Clinical Description).
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the SLC16A2 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to the sibs of a proband is much reduced, but greater than that of the general population because of the possibility of maternal germline mosaicism. Although no instances of germline mosaicism have been reported, it remains a possibility.
Offspring of a male proband. Affected males are not known to reproduce.
Other family members. The proband's maternal aunts may be at risk of being carriers (typically asymptomatic, although they may have minor thyroid test abnormalities) for the pathogenic variant and the aunts' offspring, depending on their sex, may be at risk of being carriers for the pathogenic variant or of being affected.
Note: Molecular genetic testing may be able to identify the family member in whom a de novo pathogenic variant arose – information that could help determine genetic risk status of the extended family.
Heterozygote Detection
Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the SLC16A2 pathogenic variant has been identified in the proband.
Note: (1) Females who are heterozygous (carriers) for this X-linked disorder will typically be asymptomatic, although they may have minor thyroid test abnormalities (see Clinical Description). (2) Identification of female heterozygotes requires either (a) prior identification of the SLC16A2 pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and if no pathogenic variant is identified, by gene-targeted deletion/duplication analysis.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC16A2 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MCT8-AHDS FoundationCanadaEmail: contact@mct8.info
- Una Vita Rara AHDS-MCT8 ONLUS (Italian AHDS-MCT8 Family Association)Via Foina, 3425040 Monticelli Brusati (BS)ItalyPhone: 39 329 0648896Email: unavitarara@gmail.com
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968Fax: 202-387-2193
- Association Française XtraordinaireFrancePhone: 09 70 40 61 40Email: contact@xtraordinaire.org
- CDC - Developmental DisabilitiesPhone: 800-CDC-INFOEmail: cdcinfo@cdc.gov
- EURORDIS-Rare Diseases EuropeEmail: eurordis@eurordis.org
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Allan-Herndon-Dudley Syndrome: Genes and Databases
Table B.
OMIM Entries for Allan-Herndon-Dudley Syndrome (View All in OMIM)
Molecular Pathogenesis
Monocarboxylate transporter 8 (MCT8), the protein product of SLC16A2, is thought to play a crucial role in neuronal T3 uptake and in endothelial cells allowing partial entry of thyroid hormone through the blood-brain barrier. MCT8 deficiency results in an insufficient supply of T3 to nuclear T3 receptors. Thyroid hormone plays a crucial role in brain development. Thus, it is presumed that the decreased access of T3 to brain cells can lead to the severe defects in neurologic development seen in males with AHDS [Friesema et al 2006, Roberts et al 2008, Ceballos et al 2009] and in the control of blood thyroid hormone.
Mechanism of disease causation. AHDS results from a loss of function of the MCT8 protein. Most pathogenic variants are loss-of-function variants that cause decreased activity or complete inactivation of the MCT8 thyroid hormone cell-membrane transporter [Friesema et al 2006]. Variants leading to incomplete inactivation of the protein can lead to milder phenotypes (see Table 7).
SLC16A2-specific laboratory technical considerations. SLC16A2 has two translation start sites, which generate proteins of either 613 amino acids or 539 (NP_006508.2) amino acids. The transcript encoding the 539-amino-acid protein is the one currently recognized by NCBI (Table 7).
Table 7.
Notable SLC16A2 Pathogenic Variants
Chapter Notes
Acknowledgments
For the original publication of this chapter, Drs Dumitrescu and Refetoff were supported in part by grants DR15070, DK07011, RR04999 and DK091016 from the National Institutes of Health. Dr Fu was funded by an award from China Scholarship Council.
Dr Sarret was supported by the French PHRC "XMLR." Drs Oliver and Tonduti were supported as investigators by the ERASMUS University (The Netherlands). They thank the Xtraordinaire association and patients' families for their help in conducting their research.
Author History
Melissa A Dempsey, MS; Parkview Health, Ft Wayne, Indiana (2009-2020)
Alexandra M Dumitrescu, MD, PhD; University of Chicago Medical Center (2009-2020)
Jiao Fu, MD; University of Chicago Medical Center (2013-2020)
Isabelle Oliver Petit, MD (2020-present)
Samuel Refetoff, MD; University of Chicago Medical Center (2009-2020)
Catherine Sarret, MD, PhD (2020-present)
Davide Tonduti, MD, PhD (2020-present)
Revision History
- 16 January 2020 (bp) Comprehensive update posted live
- 11 April 2013 (me) Comprehensive update posted live
- 9 March 2010 (me) Review posted live
- 6 July 2009 (mad) Original submission
References
Literature Cited
- Anık A, Kersseboom S, Demir K, Catlı G, Yiş U, Böber E, van Mullem A, van Herebeek RE, Hız S, Abacı A, Visser TJ. Psychomotor retardation caused by a defective thyroid hormone transporter: report of two families with different MCT8 mutations. Horm Res Paediatr. 2014;82:261–71. [PubMed: 25247785]
- Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663–6. [PubMed: 15834651]
- Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, Morte B, Bernal J. Importance of monocarboxylate transporter 8 (Mct8) for the blood-brain barrier dependent availability of 3,5,3'-triiodo-L-thyronine (T3). Endocrinology. 2009;150:2491–6. [PMC free article: PMC2671898] [PubMed: 19147674]
- Dumitrescu AM, Liao X-H, Best TD, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–75. [PMC free article: PMC1181904] [PubMed: 14661163]
- Dumitrescu AM, Refetoff S. Cell transport defects. In: Wondisford FE, Radovick S, eds. Clinical Management of Thyroid Disease. Chap 22. Philadelphia, PA: Elsevier Saunders; 2009:317-23.
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–7. [PubMed: 15488219]
- Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2:512–23. [PubMed: 16957765]
- Friesema EC, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. 2010;322:107–13. [PubMed: 20083155]
- Frints SGM, Lenzner S, Bauters M, Jensen LR, Esch HV, des Portes V, Moog U, Macville MVE, van Roozendaal K, Schrander-Stumpel CTRM, Tzschach A, Marynen P, Fryns J, Hamel B, van Bokhoven H, Chelly J, Beldjord C, Turner G, Gecz J, Moraine C, Raynaud M, Ropers HH, Froyen G, Kuss AW. MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet. 2008;16:1029–37. [PubMed: 18398436]
- Fuchs O, Pfarr N, Pohlenz J, Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009;51:240–4. [PubMed: 19018842]
- García-de Teresa B, González-Del Angel A, Reyna-Fabián ME, et al. Deletion of exon 1 of the SLC16A2 gene: a common occurrence in patients with Allan-Herndon-Dudley syndrome. Thyroid. 2015;25:361–7. [PubMed: 25517855]
- Gika AD, Siddiqui A, Hulse AJ, Edward S, Fallon P, McEntagart ME, Jan W, Josifova D, Lerman-Sagie T, Drummond J, Thompson E, Refetoff S, Bönnemann CG, Jungbluth H. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol. 2010;52:475–82. [PMC free article: PMC5800746] [PubMed: 19811520]
- Groeneweg S, Peeters RP, Moran C, Stoupa A, Auriol F, Tonduti D, et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019;7:695–706. [PMC free article: PMC7611958] [PubMed: 31377265]
- Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, Ramirez P, Iorcansky S, Rivarola MA, Belgorosky A. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2007;67:1–6. [PubMed: 16974106]
- Holden KR, Zuñiga OF, May MM, Su H, Molinero MR, Rogers RC, Schwartz CE. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005;20:852–7. [PubMed: 16417886]
- Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Grüters A, Barrett TG, Mancilla EE, Svensson J, Wemeau JL, Busi da Silva Canalli MH, Lundgren J, McEntagart ME, Hopper N, Arts WF, Visser TJ. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92:2378–81. [PubMed: 17356046]
- Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. 2008;149:2184–90. [PMC free article: PMC2734492] [PubMed: 18187543]
- Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. 2005;147:552–4. [PubMed: 16227048]
- Masnada S, Groenweg S, Saletti V, Chiapparini L, Castellotti B, Salsano E, Visser WE, Tonduti D. Novel mutations in SLC16A2 associated with a less severe phenotype of MCT8 deficiency. Metab Brain Dis. 2019;34:1565–75. [PubMed: 31332729]
- Novara F, Groeneweg S, Freri E, Estienne M, Reho P, Matricardi S, Castellotti B, Visser WE, Zuffardi O, Visser TJ. Clinical and molecular characteristics of SLC16A2 (MCT8) mutations in three families with the Allan-Herndon-Dudley syndrome. Hum Mutat. 2017;38:260–4. [PubMed: 27805744]
- Philips AK, Sirén A, Avela K, Somer M, Peippo M, Ahvenainen M, Doagu F, Arvio M, Kääriäinen H, Van Esch H, Froyen G, Haas SA, Hu H, Kalscheuer VM, Järvelä I. X-exome sequencing in Finnish families with intellectual disability--four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis. 2014;9:49. [PMC free article: PMC4022384] [PubMed: 24721225]
- Ramos HE, Morandini M, Carre A, Tron E, Floch C, Mandelbrot L, Neri N, De Sarcus B, Simon A, Bonnefont JP, Amiel J, Desguerre I, Valayannopoulos V, Castanet M, Polak M. Pregnancy in women heterozygous for MCT8 mutations: risk of maternal hypothyroxinemia and fetal care. Eur J Endocrinol. 2011;164:309–14. [PubMed: 21098685]
- Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab. 2007;21:277–305. [PubMed: 17574009]
- Remerand G, Boespflug-Tanguy O, Tonduti D, Touraine R, Rodriguez D, Curie A, Perreton N, Des Portes V, Sarret C, et al. RMLX/AHDS Study Group. Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev Med Child Neurol. 2019;61:1439–47. [PubMed: 31410843]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N. Expression of the thyroid hormone transporters MCT8 (SLC16A2) and OATP14 (SLCO1C1) at the blood-brain barrier. Endocrinology. 2008;149:6251–61. [PubMed: 18687783]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the MCT8 thyroid hormone transporter. Am J Hum Genet. 2005;77:41–53. [PMC free article: PMC1226193] [PubMed: 15889350]
- Schwartz CE, Stevenson RE. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab. 2007;21:307–21. [PMC free article: PMC2094733] [PubMed: 17574010]
- Sijens PE, Rödiger LA, Meiners LC, Lunsing RJ. 1H magnetic resonance spectroscopy in monocarboxylate transporter 8 gene deficiency. J Clin Endocrinol Metab. 2008;93:1854–9. [PubMed: 18319316]
- Tonduti D, Vanderver A, Berardinelli A, Schmidt JL, Collins CD, Novara F, Di Genni A, Mita A, Triulzi F, Brunstrom-Hernandez JE, Zuffardi O, Balottin U, Orcesi S. MCT8 deficiency: extrapyramidal symptoms and delayed myelination as prominent features. J Child Neurol. 2013;28:795–800. [PMC free article: PMC4155008] [PubMed: 22805248]
- Tsurusaki Y, Osaka H, Hamanoue H, Shimbo H, Tsuji M, Doi H, Saitsu H, Matsumoto N, Miyake N. Rapid detection of a mutation causing X-linked leucoencephalopathy by exome sequencing. J Med Genet. 2011;48:606–9. [PubMed: 21415082]
- Vaurs-Barrière C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Vinas JM, De Michele G, Dan B, Brady AF, Boespflug-Tanguy O, Touraine R. Pelizaeus-Merzbacher-like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65:114–18. [PubMed: 19194886]
- Visser WE, Jansen J, Friesema EC, Kester MH, Mancilla E, Lundgren J, van der Knaap MS, Lunsing RJ, Brouwer OF, Visser TJ. Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat. 2009;30:29–38. [PubMed: 18636565]
- Visser WE, Vrijmoeth P, Visser FE, Arts WF, van Toor H, Visser TJ. Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin Endocrinol (Oxf). 2013;78:310–5. [PubMed: 22924588]
- Yamamoto S, Okuhara K, Tonoki H, Iizuka S, Nihei N, Tajima T. A novel deletion mutation of SLC16A2 encoding monocarboxylate transporter (MCT) 8 in a 26-year-old Japanese patient with Allan-Herndon-Dudley syndrome. Clin Pediatr Endocrinol. 2013;22:83–6. [PMC free article: PMC3809735] [PubMed: 24170966]
- Zung A, Visser TJ, Uitterlinden AG, Rivadeneira F, Friesema EC. A child with a deletion in the monocarboxylate transporter 8 gene: 7-year follow-up and effects of thyroid hormone treatment. Eur J Endocrinol. 2011;165:823–30. [PubMed: 21896621]
Publication Details
Author Information and Affiliations
Centre Hospitalier Universitaire de Clermont-Ferrand
Clermont-Ferrand, France
Publication History
Initial Posting: March 9, 2010; Last Update: January 16, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Sarret C, Oliver Petit I, Tonduti D. Allan-Herndon-Dudley Syndrome. 2010 Mar 9 [Updated 2020 Jan 16]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

