

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Bergman NH, editor. Comparative Genomics: Volumes 1 and 2. Totowa (NJ): Humana Press; 2007.

Summary
PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) derives a position-specific scoring matrix (PSSM) or profile from the multiple sequence alignment of sequences detected above a given score threshold using protein–protein BLAST. This PSSM is used to further search the database for new matches, and is updated for subsequent iterations with these newly detected sequences. Thus, PSI-BLAST provides a means of detecting distant relationships between proteins. In this chapter, we discuss practical aspects of using PSI-BLAST and provide a tutorial on how to uncover distant relationships between proteins and use them to reach biologically meaningful conclusions.
1. Introduction
BLAST (Basic Local Alignment Search Tool) is a sequence similarity search method, in which a query protein or nucleotide sequence is compared to nucleotide or protein sequences in a target database to identify regions of local alignment and report those alignments that score above a given score threshold ( [1]; and Chapter 9). Position-Specific Iterative (PSI)-BLAST is a protein sequence profile search method that builds off the alignments generated by a run of the BLASTp program. The first iteration of a PSI-BLAST search is identical to a run of BLASTp program (1). It then generates a multiple alignment of the highest scoring pairs of the BLASTp run above a certain preset score or e-value threshold and calculates a profile or a position-specific score matrix (PSSM) from the multiple alignment. The PSSM captures the conservation pattern in alignment and stores it as a matrix of scores for each position in the alignment-highly conserved positions receive high scores and weakly conserved positions receive scores near zero. This profile is used in place of the original substitution matrix for a further search of the database to detect sequences that match the conservation pattern specified by the PSSM. The newly detected sequences from this second round of the search, which are above the specified score (e-value) threshold are again added to alignment the profile is refined for another round of searching. This process is iteratively continued until desired or until convergence, i.e., the state where no new sequences are detected above the defined threshold. The iterative profile generation process makes PSI-BLAST far more capable of detecting distant sequence similarities than a single query alone in BLASTp, because it combines the underlying conservation information from a range of related sequence into a single score matrix. In the evolution, three-dimensional (3D) structures of proteins may be conserved even after considerable erosion of their sequence similarity. PSI-BLAST has been demonstrated to be useful in detecting such relationships via sequence searches, which were previously only detected through direct comparison of the 3D structures (1,2). In this chapter, we discuss practical aspects of using PSI-BLST and provide a tutorial on how to uncover distant relationships between proteins and use them to reach biological meaningful conclusions.
PSI-BLAST is most conveniently used on the internet with the help of the graphical user interface provided by the PSI-BLAST search page on National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/BLAST/). The PSI-BLAST page may be customized by the user in terms of automated or semiautomated or “two-page formatting” and other parameters modified as desired. This page can then be saved as permanent internet bookmark for repeated use on future occasions. As a rule of the thumb, beginners are advised to use the profile-inclusion threshold of expect (e)-value = 0.005 for their analysis (see Note 1). However, a user familiar with globular domains and compositional bias may use the inclusion threshold of 0.01 for inclusion in the profile, if a sequence does not have any major compositionally biased segments (see Subheading 4 and ref. 3 for further details on compositional bias). A pair of protein sequences can either be homologous (sharing a common evolutionary ancestor) or nonhomologous (evolutionarily unrelated). It should be noted that PSI-BLAST does not offer a direct binary decision on whether two sequences are related or not. However, the e-value obtained for a PSI-BLAST alignment can be used as a guide for this purpose. As a heuristic it may be assumed that any compositionally unbiased query, encompassing a globular domain in a protein, giving a hit with e-value = <0.01 is likely to be an indication of a homologous relationship. However, a user must carefully evaluate such alignments case-by-case because there can occasionally be false-positives. A user may set the number of alignments and hits view as at least 1000 if searching the nonredundant (nr) database of NCBI, because of the large number hits obtained due to the current size of the database. PSI-BLAST may also be downloaded and run as a standalone program for Windows or UNIX-type operating systems. However, in this case the various parameters need to be specified using the set of command-line flags for the program. An advantage of using the standalone version is the ability to use alignments as queries to generate a starting PSSM, or saving and reusing the profile generated by a run of PSI-BLAST.
In the tutorial below the first example demonstrates how the structural and functional similarities between the Escherichia coli DNA polymerase III β-subunit and eukaryotic proliferating cell nuclear antigen (PCNA) can be identified and investigated using the PSI-BLAST program. In the next example, we demonstrate the strength of PSI-BLAST in exploring the function of an uncharacterized protein by means of identifying its 3D structural template. Emphasis here is chiefly on the practical steps involved, although when required, some of the relevant theoretical background is also provided.
2. Problem 1
2.1. Background
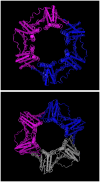
Cellular DNA polymerase enzymes tend to dissociate from DNA after adding a few nucleotides and require an accessory factor to tether them to DNA while elongating the growing DNA chain (4). In eukaryotes and archaea, this function is performed by the protein called PCNA, whereas in prokaryotes such as E. coli the same function is performed by the β-subunit of DNA polymerase encoded by the dnaN gene. When the crystal structure of E. coli DNA polymerase III β-subunit was solved, it was found to be ring shaped [5]; Fig. 1). The β-subunit forms a ring around the DNA and holds the polymerase on DNA, hence, it is also called β-clamp. It was predicted that PCNA proteins will also possess a similar ring-shaped structure (5). The crystal structure of PCNA confirms that it is similar to that of E. coli β-subunit [6,7]; Fig. 1). They all appear to have a sixfold symmetry; however, E. coli DNA Pol-III β-subunit is a dimer, where as PCNA is a trimer. Each monomer of the E. coli protein contains three homologous domains, whereas each monomer of PCNA proteins consists of two homologous domains (Fig. 1). Each domain contains an identical fold consisting of two α-helices and eight β-sheets (nine in PCNA). The proteins are negatively charged, however, two α-helices are positively charged apparently nonspecifically clamp around DNA. The sequences of β-subunit proteins are well-conserved in prokaryotes and that of PCNA in eukaryotes, but despite performing similar functions, and having some sequence conservation based on their 3D structure (5), the conventional BLASTp program detects no sequence similarity between these proteins. This distant sequence similarity, however, can be detected by PSI-BLAST as demonstrated in Problem 1. When we use human PCNA (accession number NP_002583) as the query and nr as the database, E. coli β-subunit is retrieved in the fifth iteration.
2.2. Practical Steps
- Access PSI-BLAST from the BLAST page http://www.ncbi.nlm.nih.gov/BLAST/.
- Use the default parameters (except the number of alignments and descriptions) such as “nr” as the database (see Note 3),e-value 10 and the statistical significance threshold to include a sequence for generating the PSSM for the next iteration as 0.005. Change the maximum number of alignments and descriptions, 1000, from the respective pull down menus to retrieve possibly all statistically significant hits.
- Format to get the results. The results are retrieved in another web page. The hits are divided into two sections. The hits with better statistical significance than the e-value threshold, 0.005, are listed first. Those with e-values worse than threshold, but have an e-value better than that selected on the query page, 10, are listed further down the page. Hits with e-values better than the threshold are used in forming the profile that will be used in subsequent PSI-BLAST iterations (see Note 4). It will be observed that most of the hits are to eukaryotic PCNA.
- Click on the “taxonomy reports” to get a list of hits and their organisms. These are mostly eukaryotes and archaea.
- Click the “Run 2nd iteration” button and then the “Format” link on the BLSAT page (see Note 5).

An examination of the sequence alignment of PCNA and the DNA pol-III β-subunit reveals a conservation pattern that is chiefly comprised of two types of residues:(1)The hydrophobic residues that are distributed throughout the length of the alignment and (2) polar (principally charged) residues distributed sporadically in the alignment. A comparison of the conserved positions using the structures of PCNA and the β-subunit as a guide illustrate that the conserved hydrophobic positions are those that are required for forming the hydrophobic core that folds into the interior of the protein domain and stabilizes via hydrophobic interaction. Thus, they are the critical determinants of the common fold assumed by PCNA and the β-subunit. Likewise it is seen, that the polar residues localize to the solvent or ligand-exposed surfaces of the molecule. In particular, the positively charged positions localize to the interiors of the ring structure that interact with DNA, whereas the negatively charged positions localize to the exterior surface of the ring. Thus, despite the vast sequence divergence seen between PCNA and the DNA pol-III β-subunit, we observe that not only is their relationship detected using PSI-BLAST, but also subtle patterns are picked up in the alignment that have relevance for the shared folding and functional properties of these proteins.
3. Problem 2
In this problem, we will demonstrate the strength of PSI-BLAST to assign function to an uncharacterized protein and obtain its structural template. Retrieve an entry with accession number, BAE56987, in the Entrez Protein database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Protein). The Aspergillus oryzae protein is currently identified as an unnamed protein. Perform the PSI-BLAST search as described in Subheading 2. There are a number of hits to other unnamed or hypothetical proteins and a couple of histone acetyl transferases. The “G” buttons next to the hits link to the Entrez Gene report for the genes (see Note 2). The third iteration results include a number of hits to histone acetyl transferases. One of them is a hit to an experimentally determined crystal structure, 1VHS (e ~ 10−3), from the Protein Data Bank (8,9), and this is indicated in the search results by a red “S” button next to it. The sequence alignment of the query protein to 1VHS is shown in Fig. 3.

Examination of the alignment generated by the PSI-BLAST with a protein of known structure can now be used to explore the structural and biochemical properties of the uncharacterized protein. One can visualize the 3D structure of 1VHS in the aligned region and, thus, obtain a structural template for exploring the query protein using 3D structure visualization programs. For example Cn3D, a helper application for the web browsers provided by NCBI can be used directly for this purpose (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml). Click the “S” button next to the 1VHS description, then on the arrow indicating the alignment. First, by using the alignment in conjunction with the known structure (1VHS), one can show that the conserved region detected in the uncharacterized protein is likely to adopt an α + β-fold similar to 1VHS. This is not only supported by the statistically significant e-value for the relationship, but also by the conservation of several hydrophobic residues (as in Problem 1) that are likely to assume a key role in stabilizing a hydrophobic core congruent to what is observed in the structure 1VHS. Second, it may also be noticed that the proteins detected in the previous search share a key Q/RxxGxG/A motif that is found in a loop between a strand and helix. This motif is found in a large number of proteins of the GNAT supefamily and required for binding coenzyme A (10). The conservation between the uncharacterized Aspergillus protein and 1VHS of the above motif and the positions associated with active site required to bind CoA and transfer it to amino groups suggests that the former protein is likely to function as a CoA-dependent amino-group acetyltransferase enzyme (11,12). Similar analysis using PSI-BLAST has been used to assign functions to a number of uncharacterized proteins including yeast SPT10 as a histone acetyl transferase (10).
4. Caveats to Remember While Using PSI-BLAST
There are several key caveats that need to be kept in mind while using PSI-BLAST for obtaining scientifically correct results. The first of these is the effect of compositional bias in the query sequence. Compositional bias is defined as the presence of low entropy, or low information content in a protein sequence. Typically, such a sequence may be marked by enrichment of the sequence in particular amino acids, homopolymeric stretches of a particular amino acid or presence of short-range repetitive structures such as coiled-coils or short β-helices. Such sequences as a rule assume nonglobular structures and can artificially result in high-scoring alignments with other similarly biased sequences in the database. Such relationships are typically neither biologically nor evolutionarily significant and can often mask true relationships, by preventing detection of a more subtle globular domain. For this purpose, the internet version of PSI-BLAST contains certain corrective measures: (1) filtering out of the low-complexity using the SEG program (see Note 8) and (2) using composition-based statistical correction in PSI-BLAST (13). These options are available in pull-down menu and by default composition based correction is kept on. Users are strongly advised to use these measures especially if they are searching with sequences of certain eukaryotic organisms, such as Plasmodium or Dictyostelium, whose proteins are particularly enriched in low-complexity sequence. Another caveat to keep in mind is that different queries belonging to same family or superfamily of proteins can perform differently in searches against the same database in terms of retrieving other members of that family or superfamily. Hence, it is advised that a user run PSI-BLAST from different starting points and compare the hits generated in the different searches. This acts as both a consistency check, which might help in weeding out systematic false-positives generated by a certain query and at the same time widening the horizons of newly detected sequences.
5. Notes
Note 1
The e-value is a parameter that describes the number of hits one can “expect” to see by chance when searching a database of a particular size. It decreases exponentially with the score (S) that is assigned to a match between two sequences. Essentially, the e-value describes the random background noise that exists for matches between sequences.
Note 2
The user can search for the human PCNA gene entry in Entrez Gene (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene) by using the query PCNA AND human (orgn). Entrez Gene is an NCBI resource that provides detailed information about the genes from a number of organisms and links to appropriate resources within and outside NBCI (14). It provides links to the Reference Sequence (RefSeq) entries, when available. The RefSeq database provides nonredundant and curated genomic, transcript and protein sequences for major research organisms (15).
Note 3
The protein nr database consists of conceptual translations of the coding regions annotated on GenBank/EMBL/DDBJ database and protein sequences from databases such as SwissProt and Protein Data Bank. Information about other possible databases can be obtained from http://www.ncbi.nlm.nih.gov/blast/producttable.shtml#db.
Note 4
The sequences listed on the page but with e-values worse than the threshold 0.005 can be manually selected, by checking the box, for generating the profile for next iteration. Also, the sequences already included by default for generating a profile can be manually removed by unchecking the box next to it.
Note 5
As mentioned in Note 4, the BLAST results are retrieved in another page. When clicked on the “Run nth iteration” button, the PSSM generated from the previous BLAST results is searched against the database, the original BLAST search page is refreshed with the new search, and a new request id is assigned. Thus, during several iterations of PSI-BLAST, there will be only two pages, the BLAST search page and the results page. The newly added sequences that were below the threshold in the previous search are indicated as “new” and the green dots indicate the sequences that were identified in the previous iterations.
Note 6
A stand-alone version of PSI-BLAST (ftp://ftp.ncbi.nih.gov/blast/executables/) allows the user to run the program for a chosen number of iterations or until convergence.
Note 7
The results can be formatted to obtain PSSM after any iteration, instead of the default pairwise alignment, using the “Alignment” pull down menu next to the “Format” option.
Note 8
The masking of low-complexity by the SEG filter will introduce X in place of the low complexity region or depict them in lower case depending on the user’s choice.
Since this chapter was sent for publication the NCBI BLAST site has undergone a major revampling. In the new system the values entered by the user are retained in “memory”. The user can also access multiple old results and reformat or rerun the same search by changing parameters. It is suggested that the readers familiarize themselves with the new front-end, before attempting further experiments along the lines suggested in this chapter.
References
- 1.
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [PMC free article: PMC146917] [PubMed: 9254694]
- 2.
- Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999;287:1023–1040. [PubMed: 10222208]
- 3.
- Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994;18:269–285. [PubMed: 7952898]
- 4.
- Kornberg A, Baker TA, editors. DNA Replication. W. H. Freeman; New York, NY: 1991.
- 5.
- Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. [PubMed: 1349852]
- 6.
- Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. [PubMed: 8861913]
- 7.
- Moarefi I, Jeruzalmi D, Turner J, O’Donnell M, Kuriyan J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000;296:1215–1223. [PubMed: 10698628]
- 8.
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [PMC free article: PMC102472] [PubMed: 10592235]
- 9.
- Marchler-Bauer A, Addess KJ, Chappey C, et al. MMDB: Entrez’s 3D structure database. Nucleic Acids Res. 1999;27:240–243. [PMC free article: PMC148145] [PubMed: 9847190]
- 10.
- Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that include the yeast SPT10 protein. Trends Biochem. Sci. 1997;22:154–155. [PubMed: 9175471]
- 11.
- Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley S. Crystal structure of a GCN5-related N-acetyltranferase: Serratia marcescens aminoglycoside 3-N-acetyltransefrase. Cell. 1998;94:439–449. [PubMed: 9727487]
- 12.
- Clements A, Rojas JR, Trievel RC, Wang L, Berger SL, Marmorstein R. Crystal structure of the histone acetyltransferase domain of the human PCAF transcriptional regulator bound to coenzyme A. The EMBO Journal. 1999;18:3521–3532. [PMC free article: PMC1171431] [PubMed: 10393169]
- 13.
- Schäffer AA, Aravind L, Madden TL, et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. [PMC free article: PMC55814] [PubMed: 11452024]
- 14.
- Maglott D, Pruitt K, Tatusova T. Entrez gene: a directory of genes. In: McEntyre J, Ostell J, editors. The NCBI Handbook. National Library of Medicine (US), NCBI; Bethesda, MD: 2005.
- 15.
- Pruitt KD, Tatusova T, Ostell JM. McEntyre J, Ostell J, editors. The Reference Sequence (RefSeq) Project. National Library of Medicine (US), NCBI; Bethesda, MD: The NCBI Handbook. 2005 Chapter 18.
- BLAST QuickStart: example-driven web-based BLAST tutorial.[Methods Mol Biol. 2007]BLAST QuickStart: example-driven web-based BLAST tutorial.Wheeler D, Bhagwat M. Methods Mol Biol. 2007; 395:149-76.
- IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices.[Bioinformatics. 1999]IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices.Schäffer AA, Wolf YI, Ponting CP, Koonin EV, Aravind L, Altschul SF. Bioinformatics. 1999 Dec; 15(12):1000-11.
- Simple adjustment of the sequence weight algorithm remarkably enhances PSI-BLAST performance.[BMC Bioinformatics. 2017]Simple adjustment of the sequence weight algorithm remarkably enhances PSI-BLAST performance.Oda T, Lim K, Tomii K. BMC Bioinformatics. 2017 Jun 2; 18(1):288. Epub 2017 Jun 2.
- Review Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements.[Nucleic Acids Res. 2001]Review Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements.Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Nucleic Acids Res. 2001 Jul 15; 29(14):2994-3005.
- Review Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.[Nucleic Acids Res. 1997]Review Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997 Sep 1; 25(17):3389-402.
- PSI-BLAST Tutorial - Comparative GenomicsPSI-BLAST Tutorial - Comparative Genomics
Your browsing activity is empty.
Activity recording is turned off.
See more...