

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Frank SA. Immunology and Evolution of Infectious Disease. Princeton (NJ): Princeton University Press; 2002.

Experimental evolution of influenza has identified amino acid sites that mediate escape from antibody attack. Experimental studies have also located sites that influence binding to host receptors. In this chapter, I put these experimental studies in the context of influenza structure. I also discuss how amino acid substitutions affect the kinetics of antibody binding and neutralization. These rate processes influence the fitness consequences of amino acid variants and the course of evolutionary change.
The first section provides an overview of influenza antigenicity and structure. Detailed structural information exists for hemagglutinin, the key viral surface glycoprotein. Structural analyses also describe hemagglutinin bound to its host receptor and hemagglutinin bound to antibodies. These diverse structural studies set the foundation for evolutionary analyses, allowing one to develop detailed hypotheses about the forces acting on amino acid replacements.
The second section discusses antibody escape variants, many generated in experimental evolutionary studies with controlled antibody pressure. Much of the exposed surface of hemagglutinin responds to antibody pressure with escape mutants.
The third section describes experimental studies of cell binding and receptor tropism. Ancestral lineages of influenza A in birds use an α(2,3)-linked form of sialic acid as the host receptor. Derived lineages in humans use an α(2,6) linkage. Experimental evolution studies grew a human α(2,6)-tropic form in cell culture with horse serum that binds and interferes with the α(2,6)-tropic linkage. A single amino acid change of leucine to glutamine produced an α(2,3)-tropic viral receptor. The favored amino acid, glutamine, matches that found in birds at the same site. The reverse experiment began with the avian α(2,3)-tropic form and selected for human α(2,6)-tropic binding. The avian glutamine changed to leucine, matching the amino acid found in human isolates.
The fourth section analyzes the fitness consequences of amino acid substitutions. Observed substitutions can raise or lower binding affinity for the host receptor. Natural selection of affinity may balance the kinetics of binding and the kinetics of release from the widely distributed sialic acid receptor on host cells. A few studies report the effect of amino acid substitutions on antibody binding affinity. Those studies also relate antibody binding affinity to neutralization of viruses, a measure of the reduction in viral fitness. I describe preliminary studies on the mechanisms and the kinetics by which antibodies interfere with viruses. Those details will be required to understand how amino acid substitutions alter viral fitness.
The fifth section summarizes experimental evolution studies of other pathogens.
The sixth section presents topics for future research.
13.1. Overview of Antigenicity and Structure
Influenza viruses occur as three phylogenetically distinct types (Lamb and Krug 2001; Wright and Webster 2001). Influenza A and B have eight RNA segments, and influenza C has seven segments. Influenza C occurs primarily in humans, has relatively little antigenic variation, and does not cause significant disease. Influenza B occurs naturally only in humans. By contrast, influenza A infects humans, several other mammalian species including pigs and horses, and many avian species. Influenza A has much greater amino acid sequence variability than influenza B, although type B does vary among natural isolates.
The nearly annual human epidemics of influenza A or B cause significant morbidity and mortality (Nguyen-Van-Tam 1998). The yearly outbreaks often spread widely through human populations. Epidemics lead to immunological memory against common strains (Natali et al. 1981; Wang et al. 1986; Dowdle 1999; Nakajima et al. 2000). Immunological memory creates strong selective pressure on the viruses to change antigenic properties, escape immune memory responses within hosts, and initiate new outbreaks (Wilson and Cox 1990; Cox and Bender 1995). Widespread epidemics and the strong selective pressures of host immunity cause influenza A to evolve very rapidly in humans. Individual strains often die out after a few years, replaced by antigenic variants that temporarily escape immunological memory (Bush et al. 1999).
Figure 13.1 shows the organization of the influenza A virus. Hemagglutinin (HA) and neuraminidase (NA) comprise the major surface glycoproteins.

Figure 13.1
Influenza A structure. HA and NA spikes dominate the surface and form the main antigenic regions for antibody binding. Complexes of RNA and protein form the ribonucleoproteins. Eight distinct RNA segments make up the influenza A genome. Redrawn from Lamb (more...)
HA and NA reactivities with antibodies define the subtypes of influenza A (Cox and Subbarao 2000). Fifteen different HA antigenic subtypes occur, each subtype cross-reacting relatively little with the other subtypes. Nine distinct NA subtypes are known. The designation HxNy describes an influenza subtype with HA antigenic subtype x = 1,...,15 and NA antigenic subtype y = 1,...,9.
RNA sequences differ more between antigenic subtypes than within subtypes. Sequence similarity falls below 70% between HA subtypes and rises above 80% between isolates of the same subtype (Röhm et al. 1996). Thus, broad measures of antigenic and phylogenetic distances provide similar pictures of divergence. Much antigenic diversity also occurs between different members of an antigenic subtype. At these smaller distances, antigenic measures of differentiation become sensitive to the panel of antibodies and the nature of the test.
The HA and NA genes occur on different RNA segments. The other six segments contain the remaining genes. A host infected with two different viral genotypes can produce hybrid viral progeny with reassorted genotypes (Scholtissek 1998). For example, coinfection with HxNy and HwNz could produce the hybrids HxNz and HwNy in addition to the parental types.
The H3N2 subtype that caused the "Hong Kong" pandemic of 1968 arose by reassortment of the human H2N2 subtype with avian genes. The reassorted genotype had the H3 subtype and the PB1 gene from the bird lineage and the other six segments from the H2N2 human lineage (Kawaoka et al. 1989; Cox and Subbarao 2000). Other reassortments between the major human subtypes have been documented during the past twenty-five years (Cox and Bender 1995). Reassortment between subtypes may not occur frequently, but may be important in creating novel genotypes that have the potential to spread widely through a host population, causing pandemics.
All HA and NA subtypes occur in aquatic birds, suggesting that those avian species were the original host of influenza (Webster and Bean 1998; Cox and Subbarao 2000). Widespread human epidemics have been limited to H1N1, H2N2, and H3N2, although occasional transfers of other subtypes occur from birds or mammals to humans. Pigs harbor H1 and H3, whereas horses have H3 and H7. Other mammals and nonaquatic birds occasionally become infected, but do not appear to maintain stable lineages over time.
Influenza HA and NA molecules mediate viral attachment and entry to host cells and release of progeny viral particles by budding through the membrane of infected cells (Lamb and Krug 2001). Current understanding assigns adsorption and entry functions to HA (Steinhauer and Wharton 1998) and release of progeny to NA (Colman 1998). However, the HA and NA molecules may have multiple active sites and various functions, and the different subtypes of each molecule differ significantly (Lamb and Krug 2001). With those caveats, a brief summary of structure and function follows.
HA binds to sialic acid on the surface of host cells. The HA molecule then cleaves into two parts, the terminal HA1 and the basal HA2 fragments. Cleavage exposes on the surface of HA2 a highly conserved, hydrophobic region that mediates fusion and entry via the host membrane (Wilson and Cox 1990; Skehel and Wiley 2000).
Figure 13.2 shows a diagram of the HA amino acid chain and its folding into a three-dimensional structure. The sialic acid binding site occurs near the tip of HA1. The letters A–E locate the major regions for antigenically variable amino acid substitutions, although some substitutions occur over much of the exposed surface.
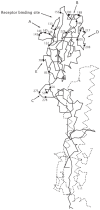
Figure 13.2
Folding of amino acid sequence to form influenza hemagglutinin HA1 (solid line) and HA2 (dotted line) subunits. This drawing is based on structural analysis of H3 hemagglutinin. Inference about other HA subtypes depends on presumed structural similarity (more...)
Figure 13.3 sketches the arrangement of the binding site and the orientation of sialic acid within the recessed contact region. The amino acids numbered within and around the binding site provide a reference for the location of important residues. The bottom of the figure shows the effect on binding affinity to sialic acid caused by experimental change of particular amino acids.
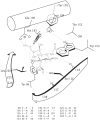
Figure 13.3
Sialic acid binding site of human influenza A hemagglutinin subtype H3. Sialic acid bound in the pocket appears in gray. The listing below shows the binding affinities for sialic acid when particular amino acids are changed experimentally by site-directed (more...)
Figure 13.4 illustrates the fit between sialic acid and the HA1 binding site. This space-filling model has roughly the same orientation as the schematic diagram in figure 13.3, allowing one to locate approximately the structures and particular amino acids shown in figure 13.3 in the spatial view of figure 13.4.

Figure 13.4
Binding of sialic acid (foreground) to the receptor binding site of influenza hemagglutinin (background). The sialic acid has been displaced slightly to show the structure of the fit. From Wilson and Cox (1990).
Antibodies bound to HA can neutralize influenza infectivity by physically obstructing the sialic acid binding site. For example, the HC19 MAb binds to HA of strain X-31 (H3 subtype), partially overlapping the sialic acid binding site (Bizebard et al. 1995). The specific antibody-epitope region of direct contact covers 1250 Å2, including amino acids 134, 136, 153, 155, and 194. Figure 13.3 shows that these residues occur on three of the four edges of the concave receptor binding depression. The depression extends 315 Å2, of which the antibody binding region covers 167 Å2. Antibody escape mutants map to the ridge of amino acids that ring the conserved amino acids in the binding pocket.
Bizebard et al. (1995) analyzed only the Fab fragments of antibodies, which contain the antibody paratope that directly binds to epitopes. Antibodies have a Y shape (fig. 2.1). Each upper arm forms an Fab fragment, with the binding region on the tip of the fragment. An antibody molecule can be cleaved to release two identical Fab fragments, each containing a binding region.
Bizebard et al. (1995) established direct overlap of the Fab binding region with the sialic acid binding site. However, other antibody escape mutants map to regions of HA away from the sialic acid binding site. Those sites are too far away to allow overlap of the direct antibody-epitope binding region with the sialic acid binding site.
Fleury et al. (1999) studied binding and neutralization by the HC45 MAb, which binds to a site distant from the sialic acid binding site. The Fab fragment of HC45 bound to its epitope with approximately the same kinetics as HC19 bound to its epitope, but HC19 was an order of magnitude more efficient at neutralization. Presumably this occurs because the Fab of HC19 causes greater obstruction of binding to sialic acid than does the more distantly bound Fab of HC45. By contrast, the full antibody molecules of HC19 and HC45 neutralized virus in proportion to their binding affinity for their respective epitopes. Fleury et al. (1999) suggest that the full antibody molecule is large enough to obstruct sialic acid binding even though the epitope binding site is distant from the sialic acid binding site.
Fleury et al. (1999) also noted a difference between HC19 and HC45 antibodies in their relative affinities for free HA molecules and HA molecules on the surface of viral particles. HC19 binds to the tips of HA molecules away from the viral surface; thus HC19 faces relatively little obstruction when binding to intact HA on viruses. By contrast, HC45 binds away from the tips of HA, toward the viral surface. This requires HC45 to diffuse through the HA spikes, slowing the rate of HA-HC45 association and reducing the net affinity of the binding. Clearly, neutralization depends on the structural environment of intact epitopes.
Other studies have noted differences between antibodies in their relations between binding and neutralization (Dimmock 1993; Schofield et al. 1997b, 1997a; Edwards and Dimmock 2000; Sanna et al. 2000). The site of antibody attachment, the kinetics of antibody binding, and the mechanism by which antibodies interfere with viral success all likely play a role in determining the strength of natural selection on various regions of the HA molecule. (See section 13.4 below, Fitness Consequences of Substitutions.)
Various structural mechanisms allow HA escape mutants to reduce binding by antibodies (Fleury et al. 1998). Bulky side chains may cause steric hindrance that interferes with antibody-epitope contact. Glycosylation adds surface carbohydrates that can prevent antibody access to potential epitopes (Caton et al. 1982; Skehel et al. 1984; Cox and Bender 1995). Some substitutions may destroy key hydrogen bonds. Alternatively, amino acid changes sometimes cause physical displacement of various protein loops.
Fleury et al. (1998) compared binding of an antibody to an original HA epitope and to a mutation of that epitope with changed conformation. When the antibody bound to the mutant epitope, the antibody-epitope complex reverted to the same structure as the antibody bound to the original type. However, the energy required to distort the conformation of the mutant epitope during binding reduced the binding affinity of the antibody by 4,000-fold relative to the affinity of the antibody for the original type. These various studies of antibody binding, structure, and kinetics provide necessary background for analyses of evolutionary change at the amino acid level.
I now turn to NA, which has not been studied as intensively as HA (Colman 1998). The function of NA is not completely understood (Lamb and Krug 2001). In general, neuraminidase enzymes cleave certain linkages within sialic acid. Sialic acid components of host cells form the primary site of influenza attachment. Thus, NA appears to cleave the host receptors to which influenza binds. This function seems to aid in releasing progeny viral particles from infected host cells.
It may be that viruses lacking neuraminidase activity enter host cells and replicate, but get stuck on the surface of the cell by attachment to sialic acid (Palese and Compans 1976). Kilbourne et al. (1968) showed that antibodies to NA do not prevent infection and replication, but do slow the rate at which viruses kill host cells—perhaps by reducing viral spread from infected to uninfected cells.
13.2. Antibody Escape Mutants
Several different experimental methods and lines of evidence can be used to infer the nature of antibody pressure on antigenic variation.
First, surface mapping determines which amino acids occur in sites accessible to antibodies. Underwood (1982, 1984) raised a panel of 125 mouse IgG MAbs against HA. Underwood compared the reactivities of the MAb panel against different natural and laboratory sequence variants of HA. Statistical methods identified which changed amino acids caused a reduction in antibody binding. The changed amino acids were located on the three-dimensional HA structure provided by Wilson et al. (1981). Almost the entire distal exposed surface of HA reacted with antibody, suggesting that the exposed regions provide a nearly continuous surface of potential epitopes. Antigenic sites B and D (fig. 13.2) contained a greater proportion of epitopes than other regions and, at least in particular laboratory mice, those sites appear to be more antigenic than other sites.
There are some problems with inferring antibody pressure by mapping surface antigenicity. Different natural and laboratory isolates of influenza may have multiple amino acid differences. This makes it difficult to assign changed antibody binding either to single amino acid substitutions or to the role of the genetic background with variations at other sites. In addition, changed antibody binding at different sites may have different consequences for binding kinetics and viral fitness. Some of the following methods mitigate these limitations.
A second approach applies MAb to either cultured or in vivo influenza (Wiley et al. 1981; Caton et al. 1982; Thomas et al. 1998). This experimental evolution favors escape variants that avoid neutralization. The locations of the escape variants map the potentially variable sites that can mutate to avoid recognition while preserving the ability to remain infectious. This antigenic map can be used to determine whether naturally varying amino acid sites likely changed under antibody pressure or by some other process.
Often, the same amino acid substitution occurs in replicate lineages faced with the same MAb, suggesting that the particular substitution provides the best balance of escape from neutralization and preservation of viral fitness. Sites that do not change under MAb pressure may either lack important contact with the antibody or may be constrained by function. These alternatives can be tested by site-directed mutagenesis, which experimentally changes particular amino acids.
A third experimental technique simultaneously applies antibodies to two or more sites (Yewdell et al. 1979, 1986; Lambkin et al. 1994). This mimics host reactions in which two or more immunodominant sites generate neutralizing antibodies. The frequency of escape mutants to a single antibody is about 10−5, so simultaneous escape against two distinct antibodies occurs at a vanishingly low frequency of 10−10. It appears that host antibodies directed simultaneously to two or more sites can greatly reduce the chance of new escape mutants during the course of a single infection.
A fourth experimental method focuses on escape mutants from low-affinity, subneutralizing antibodies (Thomas et al. 1998). Laeeq et al. (1997) obtained mice that lacked the ability to make the transition from initial, low-affinity IgM antibodies to subsequent, high-affinity IgG. They used those mice to raise low-affinity MAbs against influenza X-31 (subtype H3N2).
In previous studies, high-affinity MAbs applied to influenza typically selected single amino acid changes in one of the major antigenic sites A–E (fig. 13.2). By contrast, low-affinity MAbs selected escape mutants that had two amino acid substitutions, one in the conserved receptor-binding pocket and one in the highly antigenic regions next to the receptor-binding site.
Clearance and protection probably derive from high-affinity IgA and IgG antibodies rather than low-affinity IgM. So results from low-affinity MAbs do not reflect the most common selective pressures on antigenic variation. This study does, however, call attention to the processes by which immunodominance develops within a host. The initial, naive antibody repertoire may span widely over the HA surface, including the receptor binding pocket. The stronger antigenic sites apparently out-compete weaker sites in attracting high-affinity antibodies.
NA escape mutants have been studied less intensively than those for HA (Webster et al. 1984, 1987).
13.3. Cell Binding and Tropism
Influenza binds to sialic acid on host cells (Skehel and Wiley 2000). Sialic acid occurs as the terminal residue attached to galactose on certain carbohydrate side chains. Two common linkages between sialic acid and galactose occur in natural molecules, the α(2,3) and α(2,6) forms.
Different amino acid residues in the HA receptor binding site affect the relative affinity of HA for α(2,3) versus α(2,6) linkage (Matrosovich et al. 1997, 1998). Isolates of influenza A from aquatic birds favor the α(2,3) linkage. This matches the common α(2,3) form on the intestinal tissues of those hosts. All fifteen HA subtypes in aquatic birds share a highly conserved receptor binding site (Webster et al. 1992). The binding site apparently evolved before the evolution of the different subtypes and has been retained during subsequent divergence.
The human influenza A subtypes H1, H2, and H3 derived from avian ancestors (Webster et al. 1992). Each human subtype evolved from the matching subtype in aquatic birds, for example, human H1 from avian H1. In all three subtypes, the binding affinity of human lineages evolved to favor the α(2,6) linkage (Paulson 1985; Rogers and D'Souza 1989; Connor et al. 1994).
The evolutionary pathways differ for the human subtypes with regard to the amino acid substitutions and changes in binding that eventually led to preference for the α(2,6) form. Matrosovich et al. (1997) identified seven amino acid positions of the receptor binding site of aquatic birds that have changed during adaptation to human hosts. Human subtypes H2 and H3 have substitutions at positions 226 and 228 relative to avian ancestors. By contrast, human subtype H1 retains the ancestral avian residues at 226 and 228, but has changes in positions 138, 186, 190, 194, and 225 (see fig. 13.3 for locations of amino acid positions). Thus, different human lineages have followed different pathways of adaptation to receptor binding.
Experimental evolution studies of the H3 subtype support the phylogenetic data. Rogers et al. (1983) grew human H3N2 strains in chicken eggs in the presence of nonimmune horse serum. Horse serum contains α(2,6)-linked sialic acid, which binds to human strains of influenza and interferes with the viral life cycle. The horse serum therefore selects strongly for altered binding to α(2,3)-linked sialic acid (Matrosovich et al. 1998). After selection, Rogers et al. (1983) found a single amino acid substitution at position 226 of HA1. This substitution changed the leucine of human H3 to a glutamine residue, the same residue found in the ancestral avian H3 subtype. This substitution caused the modified virus to avoid α(2,6) binding and interference by horse serum and allowed binding to α(2,3)-bearing receptors as in the ancestral avian type.
Rogers et al. (1989) selected in the reverse direction. They began with a duck H3 isolate that had glutamine at position 226 and favored binding to α(2,3) sialic acid linkages. Binding to erythrocytes selected variants that favor the α(2,6) linkage. Viruses bound to erythrocytes were eluted and used to infect Madin-Darby canine kidney (MDCK) cells, a standard culture vehicle for human influenza isolates. This selection process caused replacement of glutamine at position 226 by leucine, which in turn favored binding of α(2,6)- over α(2,3)-linked sialic acid.
The same sort of experimental evolution on H1 isolates would be very interesting. If selection of avian H1 for a change from α(2,3) to α(2,6) binding causes the same substitutions as occurred in the human H1 lineage, then the different genetic background of avian H1 compared with H3 would be implicated in shaping the particular amino acid substitutions. By contrast, if experimental evolution favors a change at position 226 as in H3, then the evolution of human H1 receptor binding may have followed a more complex pathway than simple selection for α(2,6)-linked sialic acid.
Various steps have been proposed for adaptation of aquatic bird isolates to humans. For example, Rudneva et al. (1996) and Matrosovich et al. (1999) discuss the need to match HA and NA specificities. NA removes sialic acid from HA receptors, apparently facilitating release of viral progeny from the surface of host cells. If a viral lineage switches its HA specificity from the avian α(2,3) to the human α(2,6) form, but NA retains the avian specificity, then the lineage may have difficulty spreading in humans. Complex pathways may be required for joint adaptation of HA and NA (Matrosovich et al. 1999).
These studies raise the general problem of evolutionary pathways by which pathogens change host receptors. If two or more pathogen functions must change simultaneously, then changes in receptor affinity may be rare. The need for joint change may cause significant constraint on amino acid substitutions in receptor binding factors.
13.4. Fitness Consequences of Substitutions
Surface amino acid substitutions affect fitness in three different ways (fig. 13.5). Each of these processes relates fitness to different kinetic aspects of surface binding.

Figure 13.5
Effects of amino acid substitutions on fitness. In an experimental setting, one begins with a particular, defined genotype as the genetic background for further analysis. One then obtains single amino acid substitutions or small numbers of substitutions (more...)
First, changes in cell binding and entry affect the performance of intracellular pathogens. The relationship between binding kinetics and fitness may be rather complex. For example, figure 13.3 shows that naturally occurring amino acids may promote lower binding affinity than is associated with certain substitutions. In that figure, the substitutions 190 E→A, 225 G→R, and 228 S→G all have stronger binding affinity than the common wild type.
HA has a relatively low affinity for its host-cell receptors (Skehel and Wiley 2000). The fact that some substitutions raise affinity suggests that binding has been adjusted by selection to an intermediate rate. It may be possible to test this idea in various experimental systems by competing viruses with different cell binding kinetics.
Robertson (1993, 1999) reviews experimental evolution work on the adaptation of influenza to culture conditions in chicken eggs and Madin-Darby canine kidney (MDCK) cells. Those in vitro systems allow study of competition between different viral genotypes (Robertson et al. 1995). Simple in vitro culture conditions may select for higher binding affinity between pathogen and host cells (Robertson et al. 1995). It would be interesting to compare the fitnesses in vivo between wild type and mutants selected for higher binding affinity in vitro.
The second role of substitutions arises from binding that interferes with viral fitness. Too high affinity of HA for the primary host-cell receptor may impair release of progeny viruses. High affinity may also aggregate viruses in localized regions, interfering with infectious spread. Again, it would be interesting to compete variants with different affinities under various in vitro and in vivo conditions.
Receptor binding sites may also be strongly selected to avoid binding molecules similar to the host-cell receptor. For example, the nonimmune component of horse serum attracts influenza particles that bind the α(2,6) linkage of sialic acid (Matrosovich et al. 1998). Selection favors equine influenza strains that both bind α(2,3) linkages and avoid α(2,6) linkages. By contrast, mucins of human lungs contain α(2,3)-linked sialic acid, favoring human lineages that avoid the α(2,3) linkage (Couceiro et al. 1993). Thus, host fluids or host tissues different from the primary infection target can cull viruses from circulation. The kinetics of such fitness losses must be balanced against kinetic gains in receptor binding and avoidance of antibodies.
The third fitness effect of surface substitutions arises from changes in antibody binding. A few studies have related different aspects of antibody-virus binding kinetics to the neutralization (killing) of viruses (Schofield et al. 1997a, 1997b; Drescher and Aron 1999; Edwards and Dimmock 2000; Kostolanský et al. 2000; Sanna et al. 2000). This topic stands as a preliminary model for analyzing the relations between binding kinetics and fitness (Dimmock 1993; McLain and Dimmock 1994; Dimmock 1995).
No work has clearly established the roles of various amino acid substitutions in antibody neutralization kinetics. I highlight a few general issues and some particular studies on influenza. I suspect that experimental evolution will be an important tool in understanding the links between fitness, amino acid substitutions, the kinetics of binding to host cells, and the kinetics of antibody neutralization.
Antibody-Epitope Binding
I begin with a few key measures. Consider the simple chemical reaction [A] + [V] ⇌ [AV], where brackets denote concentration (mol/l) for antibodies, A, viruses, V, and bound antibody-virus complexes, AV. Binding occurs at the on-rate, or rate of association, ka (l/mol·s), and the breakup of bound complexes occurs at the off-rate, or rate of dissociation, kd (1/s). The equilibrium binding affinity is
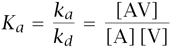
with units l/mol. At equilibrium, the binding affinities can also be given by the dissociation constant, Kd = 1/Ka.
Most studies of antibody-parasite binding report equilibrium affinity. This may capture an important aspect of neutralization, but other processes may also be important. For example, equilibrium binding affinity provides no sense of the time course of association because it describes the ratio between on-rate and off-rate. In vivo, the race occurs between the rate of antibody binding and neutralization versus the rate of pathogen attachment and entry into host cells (Dimmock 1993; McLain and Dimmock 1994). Experimental evolution studies could be devised to measure under what conditions selection favors particular changes in rate processes or only an overall change in equilibrium affinity.
Neutralization Mechanisms and Kinetics
I now turn to a few particular studies. Schofield et al. (1997a) compared equilibrium affinity, Ka, and neutralization strength for five MAbs against influenza HA. They measured neutralization by the rate at which a mixture of antibody and virus loses infectivity when presented with a layer of cultured host cells. For the five MAbs, the rank order of binding affinity approximately matched the rank order of neutralization rate. Thus, binding affinity explains some of the variation in neutralization rate. However, the ratio of affinity to neutralization rate varied by a factor of 125. Affinity alone does not explain all of the variation.
Edwards and Dimmock (2000) studied several aspects by which IgG MAbs H36 and H37 neutralize influenza. H36 binds to site B and H37 to site A on the HA molecule (see fig. 13.2). Antibodies in cell culture may neutralize by blocking viral attachment, by preventing fusion of the virus with the host cell membrane, by inhibiting internalization of the virus, or by interfering with viral replication.
Edwards and Dimmock (2000) found that, when antibodies inhibited infectivity by 50% of viruses, attachment was blocked for only 5 to 20% of viruses. Thus, other neutralizing mechanisms must play an important role. Further studies demonstrated that antibody inhibition of viral fusion increased in proportion to neutralization. Interference with fusion appears to be the primary neutralizing mechanism. However, antibody concentration influenced the relative contributions of blocking attachment versus blocking fusion: increased concentrations enhanced the degree of interference with viral attachment for both H36 and H37 antibodies. At high concentrations, interference with attachment became the dominant mechanism. As in Schofield et al. (1997a), binding affinity alone did not determine neutralization efficiency. H36 neutralized 10-fold more efficiently than did H37, but H37 binding affinity was 1.4-fold higher for H37 than for H36.
Schofield et al. (1997a) observed pseudo-first-order kinetics of influenza neutralization by antibody, defined as a log-linear decrease in infectivity over time. Pseudo-first order kinetics typically occur for antibody neutralization of viruses (Dimmock 1993), although exceptions occur (McLain and Dimmock 1994). Many different underlying mechanisms of reaction can give rise to pseudo-first-order kinetics (Latham and Burgess 1977).
The most commonly proposed mechanism for pseudo-first-order neutralization follows the single-hit model, in which one assumes that a single bound antibody can neutralize a virus (Dimmock 1993). In this model, the probability at time t that a particular virion has not been hit by at least a single antibody is e−λt, with an average time until the first hit of 1/λ. The logarithm of the number of antibody-free virions decays linearly in time with a slope proportional to −λ. This exponential decay typifies models of random waiting times, random decay, and the Poisson distribution for the number of events in a particular time period. In the antibody-virus model, one assumes an excess of antibody so that antibody pressure does not decline over time as antibodies bind to viral surfaces.
In an exponential decay model of binding, there is on average one antibody bound to each virion when λt = 1, following a Poisson distribution with an average count of one. Thus, when the average number of bound antibodies per virus is λt = 1, the single-hit model for first-order neutralization kinetics predicts a frequency of e−λt = e−1 antibody-free virions and 1 − e−1 bound and neutralized virions. Conversely, 1 − e−1 = 63% neutralization predicts an average of one bound antibody per virion.
The observed number of bound antibodies per virion at 63% neutralization varies widely (Dimmock 1993): approximately 1 for polyclonal antibodies neutralizing adenovirus hexon protein (Wohlfart 1988) and poliovirus (Wetz et al. 1986), 4 to ≥ 15 for poliovirus with MAb IgG (Icenogle et al. 1983; Mosser et al. 1989), and about 70 MAb IgG per influenza virion (Taylor et al. 1987).
First-Order Kinetics with Multihit Binding
To understand the apparent contradiction between the observed first-order kinetics and multi-hit binding, one must understand the mechanisms by which antibodies neutralize virus. Two possibilities have been discussed (Icenogle et al. 1983; Dimmock 1993; McLain and Dimmock 1994).
First, a particular epitope may occur many times on the surface of a virion. The different sites have the same antigenicity but may differ in the effect of bound antibody on neutralization. Antibody bound to critical sites neutralizes; antibody bound to noncritical sites does not neutralize. Taylor et al. (1987) found an average of 70 HA-binding IgG molecules per virion at 63% neutralization, suggesting a ratio of noncritical to critical sites of 70:1. A virion has about 1,000 HA spikes, implying about 14 critical sites per virion. Thus only one hit in 70 neutralizes. This model is possible, but at present there is no reason to suppose that only a small fraction of apparently identical HA spikes differs in some critical way.
Second, each bound antibody may partially neutralize a virion (Icenogle et al. 1983). Although this process does not yield a perfectly log-linear plot of neutralization versus time, the predicted kinetics are sufficiently close to log-linear (pseudo-first-order) that departures would not be easily noticed in experimental data. This model is attractive because a single antibody bound to one of 1,000 HA spikes on an influenza virion might fractionally reduce infectivity rather than completely neutralize the virus.
Kostolanský et al. (2000) took a different approach to understanding the interaction between the number of bound antibodies and the neutralization of influenza. Earlier studies compared different MAbs directed to different epitopes, so that it was difficult to separate the effects of the different antibodies and epitopes on the relations between affinity, neutralization kinetics, and mechanisms of neutralization. By contrast, Kostolanský et al. (2000) compared binding of a single MAb, IIB4, to different influenza strains. Those strains have variant amino acids in a single epitope located at HA antigenic site B (see fig. 13.2).
By focusing on a single MAb against variants of the same epitope, Kostolanský et al. (2000) could analyze how variations in affinity influence the number of bound antibodies and the degree of neutralization. They measured equilibrium binding affinity (Ka) of MAb IIB4 for HA variants and the ability of IIB4 to neutralize each variant. They reported neutralization as VN50, the amount of antibody in vitro required to reduce influenza replication rate by 50%.
Higher-affinity epitopes needed less antibody to reach VN50 (fig. 13.6). In addition, higher-affinity epitopes had fewer antibodies per virion at VN50. For example, the highest affinity of Ka ≈ 109 l/mol had about 13% of HA spikes occupied by antibody, whereas the lowest affinity of Ka ≈ 108 l/mol had about 98% of HA spikes occupied. These results suggest that neutralization depends on quantitative effects of affinity and the cumulative effects of multihit binding.
Figure 13.6
The amount (units in ng) of MAb IIB4 required to impose 50% neutralization (VN50) of influenza grown in cell culture. Each observation (open circle) shows the neutralization of a different influenza strain with variant amino acids at the antibody binding (more...)
The particular mechanism that leads to quantitative effects on neutralization remains unclear. It may be that lower-affinity antibodies primarily interfere with attachment to host cells by covering most viral attachment sites. By contrast, higher-affinity antibodies may interfere primarily with fusion and entry to host cells, and such steric interference at the cell surface requires a lower density of bound antibody.
Kostolanský et al. (2000) measured percent occupancy of free virions. When virions attach to cell surfaces, the lower-affinity epitopes may lose a larger fraction of bound antibody than higher-affinity epitopes. The net effect is that, to achieve VN50, both high- and low-affinity epitopes may have similar fractions of bound antibody during virion-cell binding.
Sanna et al. (2000) found that simultaneous binding by two antibodies to different epitopes acted synergistically to enhance neutralization. Synergism occurs when simultaneous binding by two antibodies causes higher neutralization than expected by adding the effects of each antibody when bound alone. Thus, the fitness effect of an amino acid substitution may depend both on the reduced affinity for the conforming antibody and on the context of other antibody-epitope combinations for that pathogen genotype.
13.5. Experimental Evolution of Other Pathogens
FMDV and influenza distinguish themselves as model experimental systems. Structural studies locate particular amino acid sites in their three-dimensional context. Experimental evolution substitutes amino acids in response to immune pressure, altered cellular receptors, interference with the viral receptor binding site, or changed kinetics that arise in cell culture. Binding affinity and kinetics of neutralization relate amino acid substitutions to components of fitness.
Several other pathogens have been studied by experimental evolution. The range of information for each pathogen tends to be limited when compared with the multiple types of evidence for FMDV and influenza. In this section, I briefly list a few additional studies of experimental evolution.
Ciurea et al. (2000) manipulated in vivo the components of the mouse immune response against cytopathic lymphocytic choriomeningitis virus. Experimental reduction of CD8+ T cell response greatly enhanced the production of escape mutants from neutralizing antibody, suggesting that antibodies play a greater part in controlling the virus in the absence of CD8+ T cells. Experimental deletion of the B cell response led to an absence of amino acid substitutions in the presumed antibody epitopes, demonstrating that substitutions in surface proteins arise in response to antibodies rather than cell tropism.
Hepatitis B virus (HBV) can cause severe liver damage in humans. Liver transplants following damage by HBV infection require suppression of HBV to prevent harm to the transplanted organ (Gow and Mutimer 2000). One suppressive method treats patients with antibody directed at the HBV surface antigens. This treatment creates an in vivo selection experiment in the patient, favoring HBV escape mutants. Not surprisingly, escape mutants do arise frequently with amino acid substitutions in the immunodominant surface antigens (Gow and Mutimer 2000).
HBV encodes surface antigens and nucleotide polymerases in different reading frames of the same nucleotide genomic sequence (Shields et al. 1999). Antigenic change in response to antibody pressure can change polymerase function, and substitutions in the polymerase in response to nucleoside analog drugs can change antigenic properties of surface proteins. The mapping of amino acid substitutions to fitness may be rather complex in this case. Ghany et al. (1998) showed that antibody escape mutants reverted to wild type after antibody pressure was removed, suggesting that the escape mutants had reduced polymerase function.
Liebert et al. (1994) studied measles encephalitis in a rat model. MAbs injected in vivo reduced neurovirulence and selected escape mutants that were isolated from brain tissue. MAb escape mutants selected in vitro produced altered and variable patterns of neurovirulence when injected into the host. The antibody epitopes appeared to be on the surface hemagglutinin protein. Amino acid substitutions in measles hemagglutinin appear to alter both antigenicity and neurovirulence.
Measles virus also appears to change its binding affinity for different cellular receptors during adaptation to cell culture (Nielsen et al. 2001). The amino acid changes associated with receptor affinity occur in the surface hemagglutinin protein. Further experimental evolution studies of this system will provide more information on how viruses modulate receptor binding and cell tropism during adaptation to different kinds of host cells.
The life cycle of arthropod-borne viruses (arboviruses) typically alternates between vertebrate hosts and blood-feeding arthropod vectors. The viruses replicate in both hosts. Arboviruses have RNA genomes and therefore the potential for high genetic diversity. However, many studies have reported a high degree of antigenic conservation and slow rates of molecular evolution (reviewed by Cooper and Scott 2001). Scott et al. (1994) and Weaver et al. (1999) suggested that alternating hosts impose a constraint on molecular change.
Cooper and Scott (2001) used experimental evolution to study how alternating hosts potentially constrain adaptive change. They passaged viral lineages in cell culture through either mosquito cells only, avian cells only, or alternating between mosquito and avian cells. They then measured various characteristics of infectivity and growth on insect, avian, or mammalian host cells.
The different passage histories produced significant differences in infectivity and growth between the lineages. The lineages that alternated between the two host types expressed intermediate phenotypes relative to those lineages passaged only in one cell type. Alternation apparently favored compromise between changing selective regimes. Further experimental evolution studies in vivo may provide more insight into how multiple selective pressures constrain the rate of evolutionary change.
Moya et al. (2000) review experimental evolution studies of vesicular stomatitis virus, an RNA virus. They particularly emphasize that high mutation rates and large population sizes of RNA viruses affect evolutionary potential by maintaining a large diversity of variant genotypes. Those variants provide material for a rapid response to new or changing selective pressures. The consequences of varying population size on the rate of adaptation have been analyzed under controlled experimental conditions.
A few bacterial studies analyzed escape mutants in response to controlled antibody pressure (e.g., Jensen et al. 1995; Solé et al. 1998). Other scattered studies of experimental evolution have been done on nonviral pathogens, but none approaches the scope of the viral experiments.
13.6. Problems for Future Research
1. Decoy antigenic variation
The first infection of a host initially stimulates the naive IgM antibody repertoire, which has relatively low affinity and broad specificity. The mature, high-affinity antibody response develops by various processes, including competition between antibodies based on binding affinity.
A pathogen gains if its most highly antigenic sites have low rates of neutralization or high rates of antigenic change. Highly antigenic decoy sites can draw antibody pressure away from sites more sensitive to neutralization or more strongly constrained against change because of essential function.
Laeeq et al. (1997) showed that IgM antibody pressure selects variants in the receptor binding site, whereas mature, high-affinity antibodies select variants in the major antigenic sites outside of the binding pocket. The immunodominant sites draw the maturing repertoire away from the binding pocket.
To what extent have immunodominant sites evolved to draw antibody pressure away from more sensitive sites? This is a difficult question, because immunodominant sites may happen to be away from receptor binding pockets or other functional sites for a variety of reasons.
No experimental systems developed so far provide a clear way to address this problem. One needs experimental control of initial antibody pressure and a feedback mechanism that enhances antibody pressure on epitopes with stronger antibody binding. Feedback favors epitopes with relatively lower rates of neutralization to evolve relatively stronger antibody binding. Such decoy sites might additionally be favored if they could tolerate a broad array of amino acid escape mutants.
This sort of experimental evolution would provide clues about the forces that have shaped immunodominance. Mathematical models of immunodominance such as those developed by Nowak and May (2000) would aid in designing experiments and clarifying evolutionary process.
2. Genetic background
Experimental evolution studies of avian and human H3 showed that a single amino acid change at position 226 of HA1 determines avian α(2,3)-tropic or human α(2,6)-tropic binding for sialic acid (Rogers et al. 1983, 1989). An open question concerns the role of the genetic background in conditioning the effects of a particular substitution. These experiments could be repeated, starting with genotypes that have different amino acid substitutions at varying distances from site 226.
3. Kinetics of pleiotropy
Several escape mutants have been generated by application of MAbs. It would be interesting to know the pleiotropic consequences of antibody escape mutants for other components of fitness, such as binding to host receptors, growth rate, and virulence. These fitness components depend on various kinetic processes within the host. A study that matched amino acid substitutions to kinetic processes would illuminate the mechanistic basis of fitness and provide insight into the microevolutionary patterns of change in proteins.
Most work on influenza has emphasized human isolates of influenza A. Those isolates can be grown in vivo in mice and other hosts, but the change in hosts compromises interpretations of kinetics and fitness. It would be interesting to develop an experimental model of influenza A in aquatic birds, the ancestral host for this virus. This would allow study of natural variation in avian isolates coupled with in vivo experimental analysis of fitness components.
4. Balancing selection of receptor binding
Influenza binding affinity for host receptors appears to be balanced at an intermediate level. Some substitutions raise affinity, and other substitutions lower affinity. Cell culture studies of FMDV and other pathogens show that binding affinity and receptor tropism evolve readily in experimental settings. It would be interesting to learn more about the selective pressures that modulate such affinities. The fitness effects no doubt depend on kinetic rates of cellular binding and entry balanced against rates of aggregation on inappropriate surfaces and in places hidden from or exposed to immune effectors. Substitutions that modulate these rate processes may also affect antibody binding. Study of these processes depends on a good in vivo system in which selective pressures can be varied and fitness components can be measured.
5. Kinetic consequences of neutralization mechanisms
Preliminary studies of neutralization kinetics provide some clues about how antibody binding affects fitness. Different mechanistic models of neutralization could be transformed into a family of mathematical models for neutralization kinetics. Those models would clarify predicted response to experimental manipulation under different assumptions, allowing better tests of the mechanistic assumptions. In addition, models would suggest how changes in different aspects of neutralization would affect viral fitness. The more sensitive steps in neutralization would be under more intense selective pressure for change, suggesting a testable prediction for which amino acid sites would be most likely to respond during experimental evolution. These studies would link molecular mechanisms, kinetic consequences, and evolutionary forces.
- Experimental Evolution: Influenza - Immunology and Evolution of Infectious Disea...Experimental Evolution: Influenza - Immunology and Evolution of Infectious Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...