

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Bast RC Jr, Kufe DW, Pollock RE, et al., editors. Holland-Frei Cancer Medicine. 5th edition. Hamilton (ON): BC Decker; 2000.


The alkylating agents and the platinum antitumor compounds form strong chemical bonds with electron-rich atoms (nucleophiles), such as sulfur in proteins and nitrogen in DNA. Although these compounds react with many biologic molecules, the primary cytotoxic actions of both classes of agents appear to be the inhibition of DNA replication and cell division produced by their reactions with DNA. However, the chemical differences between these two classes of agents produce significant differences in their antitumor and toxic effects.
Alkylating Agents
The alkylating agents were the first nonhormonal drugs to be used effectively in the treatment of cancer, and the story behind the recognition of the antitumor effects of these compounds is a remarkable one. During World War I, toxic gases were used as military weapons. The most devastating of these gases was sulfur mustard (Fig. 48.1). The compound was used as a weapon because of its vesicant effects, which produce skin irritation, blindness, and pulmonary damage. However, it was observed that troops and civilians who were exposed to sulfur mustard also developed bone marrow suppression and lymphoid aplasia. Because of these findings, sulfur mustard was evaluated as an antitumor agent.1 The closely related, but less toxic, nitrogen mustards of World War II vintage were selected for further study. Trials in patients with lymphoma demonstrated regression of tumors, with relief of symptoms.2–4 These results encouraged the search for nitrogen mustards that were more effective and less toxic and stimulated efforts to find other chemicals with antitumor activity.
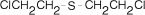
Figure 48.1
Structure of sulfur mustard (bischloroetyhylsulfide).
Chemistry of the Alkylating Agents
The alkylating agents are compounds that react with electron-rich atoms in biologic molecules to form covalent bonds. Traditionally, these agents have been divided into two types: those that react directly with biologic molecules and those that form a reactive intermediate, which then reacts with the biologic molecules. These types are termed SN1 and SN2, respectively, and are illustrated in Figure 48.2. The terms refer to the kinetics of the reactions; the rate of reaction of an SN1 agent is dependent only on the concentration of the reactive intermediate, whereas the rate of reaction of an SN2 agent is dependent on the concentration of the alkylating agent and of the molecule with which it is reacting. This distinction has important implications in understanding the cellular and molecular pharmacology of specific alkylating agents. The nitrogen mustards and nitrosoureas are examples of SN1 agents, whereas busulfan is an SN2 agent.
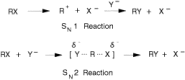
Figure 48.2
SN1 and SN2 reactions of alkylating agents.
A large number of chemical compounds are alkylating agents under physiologic conditions, and a variety of such compounds have been found to have antitumor activity. Although it is not possible to describe all of the compounds that have been used clinically, those compounds that are currently used look promising in clinical trials or represent a type of alkylating agent will be discussed.
Types of Alkylating Agents
Nitrogen Mustards
The most frequently used alkylating agents are the nitrogen mustards. Although thousands of nitrogen mustards have been synthesized and tested, only five are commonly used in cancer therapy today. These are mechlorethamine (the original “nitrogen mustard”), cyclophosphamide, ifosfamide, melphalan, and chlorambucil, and they are illustrated in Figure 48.3. The characteristic chemical constituent of the nitrogen mustards is the bischloroethyl group, and all of the nitrogen mustards react through an aziridinium intermediate as shown in Figure 48.4. The remainder of the molecule is important in determining the physical properties of the molecule and affects the transport, distribution, and reactivity of the specific agents. The importance of the total molecule is demonstrated by cyclophosphamide.

Figure 48.3
Structures of nitrogen mustards currently used in therapy.
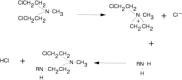
Figure 48.4
Alkylation mechanism of nitrogen mustards.
Cyclophosphamide is not a reactive compound, but it undergoes activation in the body. The complex activation scheme5 is shown in Figure 48.5. The initial activation reaction is carried out by cytochrome P450 mediated microsomal oxidation in the liver to produce 4-hydroxycyclophosphamide, which is in spontaneous equilibrium with the tautomer, aldophosphamide.6 At physiologic pH, this equilibrium is predominantly in the form of 4-hydroxycyclophosphamide.7 This equilibrium mixture diffuses from the hepatocyte into the plasma and is distributed throughout the body. Since 4-hydroxycyclophosphamide is relatively nonpolar, it enters target cells readily by diffusion. Aldophosphamide spontaneously decomposes to produce phosphoramide mustard, which is the first reactive alkylating agent produced in the metabolism of cyclophosphamide. Although phosphoramide mustard is also produced extracellularly, this compound is very polar, and enters cells poorly, and phosphoramide mustard in the plasma probably plays a minor role in the therapeutic and toxic effects of cyclophosphamide. Thus, 4-hydroxycyclophosphamide/aldophosphamide serves as an efficient mechanism to deliver the alkylating phosphoramide mustard into cells. Recent evidence suggests that after one of the chloroethyl groups of phosphoramide mustard cyclizes to form a chloroethyl azidinium moiety, the molecule cleaves to produce chloroethylaziridine.8 Accordingly, free chloroethylaziridine may contribute significantly to the alkylation and cross-linking of DNA by cyclophosphamide.

Figure 48.5
Metabolism of cyclophosphamide.
The toxic compound acrolein was demonstrated to be produced by the metabolism of cyclophosphamide by Alarcon,9 but administration of didechlorocyclophosphamide, a compound that could produce acrolein but not the chloroethyl alkylating species, did not demonstrate antitumor activity in an animal model.10 In 1992, Lee and colleagues11 reported that a decrease in the enzyme 06-alkyguanine-alkyltransferase in circulating lymphocytes was produced by the administration of high doses of cyclophosphamide for bone marrow transplantation. Recently, Friedman and colleagues reported that tumor cells with elevated 06-alkylguanine-alkyltransferase were sensitized to 4-hydroperoxycyclophosphamide by depletion of the enzyme.12 These and further studies have indicated that acrolein released by cyclophosphamide forms an 06-guanyl adduct that can be removed by 06-alkyguanine-alkyltransferase. Thus, acrolein contributes to the antitumor activity and probably the carcinogenic effects of cyclophosphamide, and these effects are abrogated by the action of O6-alkylguanine-alkyltransferase.
Cyclophosphamide produces less gastrointestinal and hematopoietic toxicity than other alkylating agents do. The basis for this decreased toxicity is the enzyme aldehyde dehydrogenase. This enzyme oxidizes aldophosphamide to carboxyphosphamide, an inactive product, which is excreted in the urine and accounts for about 80% of an administered dose of cyclophosphamide in any species. This enzyme is found in high concentration in the hepatic cytosol, in primitive hematopoietic cells, and in the stem cells and mucosal absorptive cells in the intestine.13 Administration of an inhibitor of this enzyme to an animal markedly increases the hematopoietic and gastrointestinal toxicity of cyclophosphamide.13
Ifosfamide is a structural isomer of cyclophosphamide that is used particularly in the treatment of testicular tumors and sarcomas.14–16 Ifosfamide undergoes the same metabolic reactions as cyclophosphamide, but the location of the chloroethyl group on the ring nitrogen produces quantitative changes in the metabolism of the drug17,18 and subtle changes in the chemical properties of the reactive metabolite, ifosfamide mustard, so that it is less reactive than phosphoramide mustard.19 The primary metabolite, aldoifosfamide, is a substrate for aldehyde dehydrogenase, so that the bone marrow and gastrointestinal tract sparing properties are similar to those of cyclophosphamide. The oxidation of the chloroethyl side chains to produce choroacetaldehyde is a minor metabolic pathway for cyclophosphamide (<10% of dose) but is increased to as much as 50% for ifosfamide. The increased production of chloracetaldehyde has been implicated in the neurotoxicity of ifosfamide20 and may also contribute to the greater renal and bladder toxicity of ifosfamide. The greater side chain oxidation of ifosfamide and the lesser reactivity of the ifosfamide mustard are consistent with the fact that higher doses of ifosfamide than cyclophosphamide are used clinically.
Melphalan is an alkylating agent that has been used extensively in the treatment of multiple myeloma,21,22 ovarian cancer,23,24 and breast cancer.25,26 Melphalan is an amino acid analogue that has been shown to enter cells and cross the blood-brain barrier through active transport systems. The natural substrates for these systems are amino acids,27,28 and the entry of melphalan into cells29 and the central nervous system (CNS)30 can be modulated by the presence of certain amino acids in the extracellular fluid.31
Chlorambucil has been used extensively for the treatment of chronic lymphocytic leukemia,32,33 ovarian carcinoma,34,35 and lymphoma,36,37 but it has been used less often in high-dose combination therapies than the other nitrogen mustards that are described here. This agent is well tolerated by most patients and can be used in patients who have severe nausea and vomiting with cyclophosphamide or melphalan.
Aziridines and Epoxides
Closely related to the nitrogen mustards are the aziridines, which are represented in current therapy by thiotepa, mitomycin C, and diaziquone (AZQ), illustrated in Figure 48.6. These agents presumably alkylate by the same mechanism as the aziridinium intermediates produced by the nitrogen mustards, but the aziridine rings in these compounds are uncharged and less reactive than aziridinium compounds.
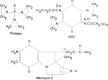
Figure 48.6
Structures of aziridine alkylating agents.
Thiotepa (triethylene thiophosphoramide) has been used particularly in the treatment of carcinomas of the ovary and breast and for the intrathecal therapy of meningeal carcinomatosis.38–40 Thiotepa is oxidatively desulfurated by hepatic microsomes to produce TEPA.41 Although TEPA is cytotoxic, it is less so than thiotepa.42 After the clinical administration of thiotepa, both thiotepa and TEPA are found in the blood,43,44 and the concentration and area under the curve (AUC) exposure to TEPA may exceed those of thiotepa.45 The AUC exposure to thiotepa has been shown to correlate with the degree of myelosuppression in patients, whereas the AUC exposure to TEPA did not.45 Some studies have suggested that a metabolite is produced that is more reactive than the parent compound.46,47 However, such a metabolite has not been characterized, and the activity of thiotepa may be enhanced by low pH within tumor cells. At the lower pH, the aziridine ring will be protonated and more reactive.
Mitomycin C is a natural product that has been used in the treatment of breast cancer and cancers of the gastrointestinal tract .48–50 This compound contains an aziridine ring and appears to exert its cytotoxic effect through the cross-linking of DNA.51,52 Mitomycin C undergoes reduction in the cell, with enhancement of the affinity of the carbon-1 atom of the aziridine ring for nucleophiles, such as the extracyclic nitrogen atom on guanylic acid in DNA. Following this alkylation, there is displacement of the activated carbamate group on the 10 carbon atom of mitomycin C by an extracyclic amino nitrogen of a guanylic acid molecule on the complementary DNA strand to produce an interstrand DNA cross-link.53–55
AZQ was designed to be sufficiently lipophilic to readily cross the blood-brain barrier for the treatment of CNS tumors.56 It has demonstrated clinical activity against brain tumors,57 other solid tumors, and leukemia.58 AZQ has been shown to undergo reduction of the quinone ring in cells. This reduction results in protonation of the aziridine rings and enhancement of reactivity of the compound.59,60
The epoxides, such as dianhydrogalactitol61,62 (Fig. 48.7), are chemically related to the aziridines and alkylate through a similar mechanism of attack of a nucleophile, such as an amino nitrogen, on a carbon of a strained three-member ring. Dibromodulcitol63 is hydrolyzed to dianhydrogalactitol and thus is a pro-drug to an epoxide.64
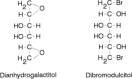
Figure 48.7
Structures of an epoxide alkylating agent (dianhydrogalactitol) and an epoxide prodrug (dibromodulcitol).
Alkyl Sulfonates
The alkyl alkane sulfonate busulfan (Fig. 48.8) was one of the earliest alkylating agents.65 This compound is one of the few currently used agents that clearly alkylate through an SN2 reaction, as shown in Figure 48.9. Hepsulfam, an alkyl sulfamate analogue of busulfan with a wider range of antitumor activity in preclinical studies,66 has been evaluated in clinical trials but thus far has demonstrated no superiority to busulfan. Busulfan has a most interesting, but poorly understood, selective toxicity for early myeloid precursors.67,68 This selective effect is probably responsible for its activity against chronic myelocytic leukemia (CML).69,70

Figure 48.8
Structure of alkyl sulfonate (busulfan) and alkyl sulfamate (hepsulfam) agents.

Figure 48.9
Mechanism of alkylation by busulfan.
The use of busulfan as first-line therapy for the treatment of CML has been succeeded by the use of the less toxic hydroxyurea. The current major use of busulfan is as a component of bone marrow ablative regimens for bone marrow and stem cell transplantation of patients with acute myeloid leukemia and other malignancies.71,72
Nitrosoureas
The nitrosoureas are a class of alkylating agents that have received considerable attention during the past 3 decades.73–75 Several nitrosoureas currently in clinical use or clinical trials are shown in Figure 48.10. These compounds decompose to produce alkylating compounds under physiologic conditions. Although there are several mechanisms by which this may occur, the predominant mechanism is that shown in Figure 48.11, a base catalyzed decomposition to a chloroethyl diazonium moiety,76 which has been shown to react with DNA,77,78 as discussed below.
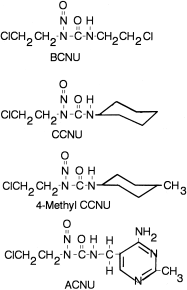
Figure 48.10
Structures of nitrosoureas.
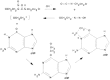
Figure 48.11
Mechanism of nitrosourea activation and alkylation of deoxyguanylic acid.
Carmustine (BCNU) was the first agent to demonstrate significant activity against a preclinical model of intracerebral tumor74 and is currently used for the treatment of primary brain tumors79 and in the treatment of multiple myeloma.80 Lomustine (CCNU) and Semustine (methyl CCNU) demonstrated greater activity against solid tumors in preclinical studies.81 CCNU is used in the treatment of CNS tumors82,83 and lymphomas,84,85 and methyl CCNU has been used particularly in the treatment of gastrointestinal tumors.86,87 ACNU, which is more water soluble than most of the other nitrosoureas, has been employed for the intra-arterial and intrathecal treatment of CNS tumors88,89 and the treatment of solid tumors.90 The clinical use of the nitrosoureas has been limited by marked and prolonged hematopoietic toxicity and by renal toxicity. The development of nitrosoureas with a higher therapeutic index, such as fotemustine91,92 and others,93,94 remains a very active area of endeavor.
Triazenes, Hydrazines, and Related Compounds
These are nitrogen-containing compounds that spontaneously decompose or can be metabolized to produce alkyl diazonium intermediates that alkylate biologic molecules. Procarbazine and dacarbazine, which are illustrated in Figure 48.12, are metabolized to reactive intermediates that decompose to produce methyl diazonium, which methylates DNA.95 The metabolism of procarbazine is complex, and there are different pathways through which a reactive methyl group can be produced.95 It is most likely that the pathway responsible for the DNA methylation and cytotoxicity is the generation of methylazoxyprocarbazine.96,97 The activation of dacarbazine via N-methyl oxidation by a microsomal P450 enzyme is illustrated in Figure 48.13.98,99 Both procarbazine and dacarbazine are used in the treatment of Hodgkins disease100,101; procarbazine is a component of combination regimens used for the treatment of primary brain tumors,102 and dacarbazine is used in the treatment of melanoma.103,104 Procarbazine was originally developed as a monoamine oxidase inhibitor, and it can produce CNS depression and acute hypertensive reactions after the ingestion of tyramine-rich foods.105
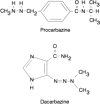
Figure 48.12
Structures of monofunctional alkylating agents.
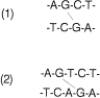
Figure 48.13
Interstrand cross-linking of DNA by nitrogen mustards. (1) Site of crosslinking proposed by Brookes.(2) Site of crosslinking found by Loechler and Hopkins.
Temozolomide (see Fig. 48.13) spontaneously decomposes under physiologic conditions to produce the same active metabolite produced by DTIC.106 Temozolomide, which is administered orally, has demonstrated antitumor activity against gliomas and melanomas in phase I and II trials107–109 and is now approved for glioma treatment in the United States and Europe.
Hexamethylmelamine
Hexamethylmelamine (Fig.48.14) is an active antitumor agent that has been considered to be acting as an alkylating agent because the methyl groups are required for antitumor activity. The methyl groups are hydroxylated with subsequent demethylation in vivo,110,111 a reaction that can generate a reactive methyl group. Analogues in which the methyl groups are hydroxylated are also active.112,113 Few studies of the cross-resistance of this agent have been carried out, but one study114 found that O-6-alkylguanine-alkyltransferase was not inactivated in vivo by hexamethylmelamine, as would be expected from an O-6 guanyl methylating agent. Therefore, the mechanism of cytotoxic activity of hexamethylmelamine remains in question. The agent does have significant antitumor activity against ovarian cancer115 and is used primarily in the third-line treatment of that tumor.

Figure 48.14
Mechanisms of resistance to alkylating agents.
Decomposition and Metabolism
The alkylating agents react with water and are inactivated by this hydrolysis. The alkylating agents also are inactivated by reaction with thiols, such as glutathione. The reaction of alkylating agents with glutathione can be increased by the glutathione S-transferase enzymes, as will be discussed below in mechanisms of cellular resistance. The alkylating agents also undergo microsomal and other types of xenobiotic metabolism. Such metabolism may activate agents, as described above, inactivate them, or change their physical properties without inactivating them. Nitrosoureas have been reported to be denitrosated and inactivated by microsomal metabolism.116,117
Chlorambucil is metabolized to bischoroethylphenylacetic acid, which is an active alkylating agent, and probably contributes to the therapeutic and toxic effects of chlorambucil.118,119 Mitomycin C must be reductively activated intracellularly to alkylate DNA bases and cross-link the DNA, and glutathione appears to play a role in this process.120
Mechanism of Cytoxicity
Although the alkylating agents react with a number of biologic molecules, including amino acids, thiols, RNA, and DNA, a number of lines of evidence have led to the generally accepted conclusion that the cytotoxic effects of the agents are due to reactions with DNA. Bifunctional agents are much more effective antitumor agents than monofunctional agents, but addition of more than two alkylating groups does not further increase the cytotoxic activity. These observations121 and the early studies of Brookes and Lawley122,123 led to the suggestion that interstrand cross-linking of DNA was responsible for the cytotoxic activity of the bifunctional alkylating agents. A good correlation has been shown between cytotoxicity and the formation of interstrand cross-links by bifunctional alkylating agents. The alkaline elution technique developed by Ewis and Kohn 124 has been especially important in these studies. More recently, nitrogen mustard interstrand cross-links in oligonucleotides have been chemically characterized.125–127
Although the alkylating agents can react with virtually all of the nitrogens in the DNA bases, there is selectivity, based on the electron density of the nitrogens and the local structure of the DNA. The nitrogen mustards react most readily with the N-7 position of guanylic acid.128 This nitrogen atom has a high electron density, which has been proposed to be enhanced by base stacking in the DNA helical structure.122 Brookes and Lawley suggested that the nitrogen mustard cross-link in DNA was between the N-7 guanine atoms in base-paired G-C sequences in DNA.122 However, more recent studies have found the nitrogen mustard cross-link to occur between the N-7 atoms of guanylic acids in a G-X-C sequence, as illustrated in Figure 48.15.125–127 The cross-linking of mitomycin C between two extracyclic guanylic acid amino groups is described above.53 This site of cross-linking may be determined by the orientation of mitomycin C in the minor groove of DNA.54 The reactive species of the nitrosoureas is more reactive than the aziridiniums of the nitrogen mustards and initially alkylates the 0-6 position of guanylic acid.129,130 According to a mechanism proposed by Ludlum, after a series of rearrangements involving a reactive cyclic five-membered intermediate of the N-1, C-6, and O-6 atoms of guanylic acid and two carbons from the chloroethyl group of the nitrosourea (see Fig. 48.11), a cross-link is formed between N-1 of guanylic acid and N-3 of a cytidylic acid on the complementary DNA strand.131,132 Such a cross-link has been demonstrated to occur in an oligonucleotide treated with BCNU.133
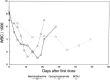
Figure 48.15
Hematopoietic toxicity of alkylating agents.
Alkylating agents such as methylnitrosourea, procarbazine, and dacarbazine are not bifunctional and produce methylation of DNA, predominantly on the O-6 and N-7 positions of guanylic acid. These lesions can produce both spontaneous and enzyme mediated single-strand breaks,134,135 which are cytotoxic. However, it has now been demonstrated, initially by Modrich and colleagues,136,137 that active mismatch DNA repair is a major mediator of the cytotoxicity of monofunctional alkylating agents.
Cellular Resistance to Alkylating Agents
Cellular resistance to antitumor agents is a critical determinant of the effectiveness of therapy. Resistance mechanisms in normal tissues provide selectivity and an improved therapeutic index. Resistance of tumor cells allows these cells to escape the effects of therapy. Consideration of the pharmacology and chemistry of the alkylating agents predicts four general types of cellular resistance to alkylating agents (Fig. 48.16). These are (a) decreased uptake of agents into or increased export out of the cell, (b) increased inactivation of agents in the cell, (c) enhanced repair of the DNA damage produced by the alkylating agents, and (d) the absence of cellular mechanisms that produce cytotoxicity in response to DNA damage. All four of these mechanisms have been described. Resistance of tumor cells to mechlorethamine can occur on the basis of decreased transport into the cell,138,139 and it has also been demonstrated that certain cells resistant to melphalan have decreased active transport of the agent and of amino acid.140,141 Most alkylating agents enter cells by diffusion, however, and the alkylating agents, with the exception of mitomycin C, are not substrates for the multiple drug resistance export systems.

Figure 48.16
Relationship between plasma AUC of busulfan and occurrence of veno-occlusive disease of the liver. (From Grochow et al.
The second mechanism of cellular resistance to alkylating agents is intracellular inactivation of the agent. As discussed above, the enzyme aldehyde dehydrogenase detoxifies the primary metabolites of cyclophosphamide and ifosfamide, and the presence of this enzyme in bone marrow precursor cells and gastrointestinal epithelial cells protects these organs from toxicity of the agents. Aldehyde dehydrogenase has also been demonstrated to be a mechanism of cyclophosphamide resistance of murine,142 rat,143 and human leukemia cells144 and human ovarian,145 colon,146 and breast147 cancer cells.
An association between cellular resistance to alkylating agents and increased cellular levels of glutathione148–150 and the enzyme glutathione transferase has been described by a number of investigators.151–154 Glutathione (GSH) is a thiol-containing tripeptide that is present at millimolar concentrations in many cells, reacts with electrophilic (electron-deficient) molecules, and protects cells from such electrophiles.155–157 Mulcahy and colleagues158 have demonstrated that increased GSH in cells resistant to melphalan can be related to increased transcription of gamma-glutamylcysteine synthetase, the enzyme that catalyzes the rate-limiting step in de novo synthesis of GSH.
Although most electrophiles of biologic significance react spontaneously with glutathione, glutathione S-transferases catalyze the reaction between glutathione and electrophiles. The glutathione conjugates of several alkylating agents have been characterized159–161 and their formation shown to be enhanced by a glutathione S-transferase.
There are three principal isozymes of glutathione S-transferase, and recent studies indicate that specific isozymes may catalyze the conjugation of different alkylating agents. The alpha isozyme of GST has been found to catalyze the glutathione conjugation of the aziridinium forms of melphalan,162 chlorambucil,163 and phosphosphoramide mustard.164 The GSH conjugation of 4-hydroxycyclophosphamide165 was found to be enhanced by all three classes of GST. The mu isozyme has been implicated in the inactivation of BCNU.166 At this time it, is evident that glutathione alone or glutathione plus an appropriate glutathione S-transferase can render cells resistant to alkylating agents and that this mechanism is probably an important mechanism of resistance to electrophilic antitumor drugs, such as the alkylating agents and platinum compounds.
Several investigators have demonstrated that buthionine sulfoxime (BSO), an inhibitor of glutathione synthesis, can reduce cellular glutathione levels and sensitize tumors to alkylating agents in vitro and in vivo.167–169 However, normal cells can also be sensitized by BSO administration170,171 to produce significant toxicity. BSO in combination with alkylating agents is now undergoing clinical trials. Phase I trials of the combination of BSO and melphalan have demonstrated increased myelotoxicity and depletion of tumor GSH, compared with the same dose of melphalan alone.172–174 Phase II trials are in progress to determine if the tumor response rate is greater with the addition of BSO to melphalan.
Inhibitors of glutathione S-transferases have been shown to enhance the cytotoxicity of melphalan on cells resistant to alkylating agents,175 and such inhibitors are being examined in clinical trials. A phase I trial of the GST inhibitor sulfasalazine176 with melphalan doses of 20 mg/m2 and greater demonstrated reductions of glutathione and GST levels in the peripheral mononuclear cells of some patients, and the main toxicity of the combination was nausea and vomiting. Increased myelosuppression was not seen.
An association between increased cellular concentrations of metallothionein and resistance to platinum agents has been established177,178 and is probably due to binding of the platinum agents to the multiple thiol groups of this cellular protein. Lazo and colleagues179 found that transfection-induced increased cellular metallothionein also produced resistance to alkylating agents. Yu and colleagues and Wei and colleagues have demonstrated binding of melphalan180 and phosphoramide mustard181 to thiol groups in metallothionein. Thus, increased metallothionein content of cells is another mechanism of inactivation of alkylating agents.
Since the cytotoxicity of the alkylating agents appears to be mediated through the alkylation of DNA, the repair of alkylation lesions is an obvious mechanism of resistance to these agents and has been the subject of intense investigation. The best-defined DNA repair resistance to alkylating agents is resistance to the nitrosoureas and other compounds that alkylate the 0-6 position of guanylic acid in DNA. The protein 0-6-alkylguanine-DNA-alkyltransferase (O6-AT) has been shown to remove alkyl groups from the 0-6 position of guanine and thus prevent the formation of an interstrand cross-link.129,131,182 The removed alkyl group is covalently and irreversibly bound to the alkyltransferase so that the protein can catalyze the removal of only one alkyl molecule and is then rapidly catabolized. It is now obvious that elevated O6-AT is a major mechanism of resistance to nitrosoureas in human gliomas183–185 and other human tumors.186–188
The fact that O6-AT is irreversibly inactivated by the transfer to it of an alkyl group from the 0-6 position of guanine provides an approach to counteracting this mechanism of resistance. If cells are treated with a monofunctional 0-6 alkylating agent, such as streptozotocin, there follows a period when the 0-6 alkyltransferase activity is decreased. This decrease in activity is due to the removal of alkyl groups from the 0-6 guanine sites on the DNA and a subsequent reduction of the level of active enzyme before enzyme synthesis can restore functional levels of the enzyme. If the cells are treated with a nitrosourea (or other 0-6 guanine alkylating agent) during this period of decreased 0-6 alkyltransferase, the cells are more sensitive to nitrosourea.189 The enzyme will also remove 0-6 benzyl groups from acid-soluble guanine analogs, and compounds such as 0-6 benzylguanine, administered prior to nitrosoureas, will reverse alkyltransferase resistance in cells and animal models,190–192 and clinical trials with 0-6 benzylguanine are now in progress.193–195
Although O6-BG and related compounds can reverse tumor resistance to nitrosoureas and methylating agents such as temozolomide, the bone marrow toxicity of these agents is increased by O6-BG. However, human O6-AT enzymes that are resistant to the combination of an alkylating agent and O6-BG have been isolated and transfected into cell lines.196,197 It has been shown in a mouse model that transfection of such an enzyme into hematopoietic cells in mice produces protection of the bone marrow from the cytotoxicity of the combination of BCNU and O6-BG,196 and currently similar studies are planned in patients.
Removal of interstrand cross-links from DNA in cells can be shown to occur in studies using alkaline elution and other techniques.198 Friedman and colleagues have described a human medulloblastoma cell that is resistant to cyclophosphamide on the basis of repair of the interstrand cross-links.199 This cell does not appear to repair the BCNU or busulfan cross-link, suggesting that the recognition and repair of interstrand cross-links are quite structure specific.
Recently, evidence has been presented that poly(ADP ribose)polymerase is involved in the repair of nitrogen mustard lesions.200 Also, there is good evidence that cells that react to alkylation damage by arresting in the G2 phase of the cell cycle can repair DNA during this period and are more resistant to alkylating agents than cells that proceed through mitosis despite alkylation damage. A human tumor cell line has been described that exhibits G2 arrest in response to alkylating damage and demonstrates increased resistance to nitrogen mustard.201 This cell line was found to have increased accumulation of phosphorylated (and inactivated) cdc2 kinase associated with G2 arrest after nitrogen mustard treatment. This alteration should allow repair of DNA damage before the cell enters mitosis. This mechanism of resistance to alkylating agents is probably important for tumor cells but also may provide a degree of drug specificity for many other tumors, because normal cells may be more likely to exhibit this protective mechanism. Inhibitors of DNA repair have been shown to enhance the cytotoxicity of alkylating agents,202–204 and some of these inhibitors are being examined in clinical trials. It seems likely that increased understanding of the DNA repair process will allow more effective use of alkylating agents.
In Vivo Resistance
Murine tumors that are resistant to alkylating agents in vivo, but not in vitro, have been reported.205,206 Further studies of these tumors that are resistant to cyclophosphamide, cisplatin, and thiotepa in vivo have demonstrated that the tumors are also resistant to these agents in three-dimensional in vitro culture but not in two-dimensional in vitro culture.207 Such resistance may be acquired rapidly after drug exposure208 and may be associated with enhanced metastatic properties.209 The mechanisms responsible for this type of resistance have not yet been established. There may be differences between known cellular resistance factors or between membrane properties in the three-dimensional milieu, compared with the two-dimensional configuration, and adhesion molecules may alter drug sensitivity. Other potential mechanisms for drug resistance in vivo are poor perfusion of the tumor and changes in the intracellular pH.210
Clinical Pharmacology
Cyclophosphamide
After the administration of a systemic dose of 50 mg/kg, plasma levels of the parent compound of up to 400 micromolar may be achieved and decay with a half-life of 3 to 10 hours.211–213 The rate of metabolism of the parent compound varies considerably among individuals and can be modulated by the administration of compounds that affect the rate of microsomal metabolism, such as phenobarbital214 or a previous dose of cyclophosphamide.215,216 However, at conventional doses, the clearance rate of the parent compound does not appear to significantly affect the toxicity or therapeutic effect of the agent.217 This independence of effect from the rate of metabolism is probably because the parent compound is not rapidly excreted and continues to be activated, so that the AUC for systemic exposure to the active metabolites is similar after a given dose.
At the higher doses currently used in bone marrow transplantation regimens, however, the plasma concentrations of cyclophosphamide should be close to the capacity of the microsomal activating enzymes. Grochow and colleagues218 demonstrated that in patients receiving 4 g/m2 of cyclophosphamide over 90 minutes and achieving initial plasma concentrations of greater than 500 mM, saturable pharmacokinetics are seen. These investigators concluded that when the dosing rate equals or exceeds 4 g/m2 in 90 minutes or the plasma concentration of cyclophosphamide exceeds 150 mM (the lowest Km seen in the patients), nonlinear disposition may occur, with variable exposure to the active metabolites. This study also confirmed previous reports that cyclophosphamide can induce its own metabolism.
Studies of pharmacokinetics of the critical metabolite 4-hydroxycyclophosphamide have been limited in the past by the difficulty of accurately measuring this labile molecule. However, more specific methods are now available, and the pharmacokinetics of this important metabolite are now being elucidated. Anderson and colleagues measured 4-hydroxycyclophosphamide in patient blood after cyclophosphamide administration, using a very specific gas chromatographic-mass spectrometric technique.219 After a dose of cyclophosphamide of 110 mg/kg over 90 minutes, peak concentrations of 9 to 12 micromolar and AUCs of 105 to 110 micromolar hours were measured; a cyclophosphamide dose of 170 mg/kg given as a continuous infusion over 4 days produced plasma concentrations of 1 to 5 micromolar, with a total AUC of about 98 to 110 micromolar hours. Subsequent studies have found similar results.220,221 All studies have found a considerable patient variation in the exposure to 4-hydroxycyclophosphamide after the same dose of cyclophosphamide and differences in the exposure and ratios of cyclophosphamide/4-hydroxycyclophosphamide each day when short-duration infusions are given on subsequent days. These findings are most likely to be due to differences in the cytochrome p450 complements in patients and the differing exposures to drugs that modulate the activities of these enzymes. These findings indicate that pharmacokinetically guided dose adjustment will be the best method to produce consistent patient exposures to the active metabolites of cyclophosphamide.
The majority of a dose of cyclophosphamide (< 70%) is excreted in the urine as the inactive metabolite carboxyphosphamide.222,223 Renal function does not significantly affect the toxicity of cyclophosphamide,224 most likely because spontaneous decomposition, and not renal excretion, determines the clearance of the principal active metabolites.
The clinical pharmacology of ifosfamide has been much less studied but is similar to that of cyclophosphamide, except that microsomal activation is somewhat slower, and chloroethyl side chain oxidation plays a greater role in its metabolism.225–227 Thus, for a dose of ifosfamide, lower systemic concentrations of the 4-hydroxy metabolite are achieved than for the same dose of cyclophosphamide.228 Both cyclophosphamide and ifosfamide are well absorbed after oral administration.229 Boddy and colleagues17 have demonstrated that ifosfamide, like cyclophosphamide, can autoinduce its own metabolism. Because of the greater and more variable side chain oxidation of ifosfamide, differences in the P450 drug metabolizing enzymes between individuals, and the modulation of these enzymes by concomitantly administered agents, may play a greater role in altering the clinical effects of ifosfamide than cyclophosphamide.230–232
Melphalan
Alberts and colleagues found that peak plasma levels of 4 to 13 micromolar were present after intravenous administration of a 0.6-mg/kg dose of melphalan, and the half-life (t1/2b) was 1.8 hours.233,234 At this dose, the mean AUC for melphalan was 8 micromolar hours. Similar AUC per dose and pharmacokinetics have been demonstrated by other investigators after high intravenous doses of melphalan.235 After conventional oral doses of 0.25 mg/kg, peak plasma levels of up to 0.625 micromolar were found.236 There is variable systemic availability after oral dosing,237,238 and it has been shown that oral administration of food with melphalan will inhibit absorption of the agent.239 It has been reported that myelosuppression from melphalan is increased in patients with decreased renal function.240 The half-life of melphalan is prolonged in anephric dogs,241 and significant renal clearance of the parent compound in patients has been shown by Reece and colleagues.242
Chlorambucil
After the oral administration of 0.6 mg/kg of chlorambucil, peak levels of 2 to 6 micromolar parent compound were found at 1 hour by Alberts and colleagues118,234 Peak plasma levels of phenylacetic acid mustard of 2 to 4 micromolar occurred at 2 to 4 hours after chlorambucil administration. The plasma half-life (t1/2b) of chlorambucil was 92 minutes and that of phenylacetic acid mustard was 145 minutes. At a dose of 0.6 mg/kg of chlorambucil, the plasma AUC of chlorambucil was 3 to 9 micromolar hours.118 Similar values were found by Hartvig and colleagues,243 who also found a two- to four-fold variation in systemic availability of chlorambucil and phenylacetic acid mustard after oral administration of chlorambucil.
Thiotepa
The pharmacokinetics of thiotepa have been studied by Cohen and colleagues,244 after an intravenous injection of 12 mg/m2. Peak plasma levels of about 5 micromolar were achieved and were found to decay with a t1/2 a of 7.7 minutes and a t1/2b of 125 minutes. The mean AUC was 9 micromolar hours. Plasma concentrations of TEPA of up to 1 micromolar were found and remained in plasma longer than thiotepa. Henner and colleagues245 examined the plasma levels of thiotepa after 4-day continuous intravenous infusions of up to 900 mg/m2. Peak plasma levels of thiotepa of 7 micromolar were initially attained on the first day, and then the levels gradually decreased. Plasma AUC values of up to 600 micromolar hours were achieved. When given intraperitoneally, there is rapid loss of thiotepa from the intraperitoneal cavity and a concomitant increase in plasma levels to those associated with the same dose if given intravenously.246 After intravenous injection, cerebrospinal fluid levels comparable with plasma levels are found.247 Recent studies have indicated that the simultaneous administration of thiotepa and cyclophosphamide will result in lower exposure to the active metabolite of cyclophosphamide, 4-hydroxycyclophosphamide.218
Nitrosoureas
The pharmacokinetics of BCNU have been studied by Levin and colleagues248 After intravenous infusion of 60 to 170 mg/m2, peak plasma concentrations of 5 micromolar were reached and then decayed with an initial half-life of 6 minutes and a second half-life of 68 minutes. Henner and colleagues249 measured the pharmacokinetics of BCNU after intravenous doses of 600 mg/m2. The peak plasma level of ultrafilterable BCNU was found to be 4.7 micromolar and the mean AUC was 5.4 micromolar hours. The ultrafilterable BCNU was 23% of the total plasma BCNU. The pharmacokinetics of CCNU after administration of 130 mg/m2 to patients have also been described.250 The parent compound could not be detected in plasma, but the monohydroxylated metabolites, trans-4-hydroxy CCNU and cis-4-hydroxy CCNU, were found in a ratio of 6:4 and at total peak concentrations of about 3 micromolar. The plasma clearance half-lives of the hydroxy-CCNU metabolites varied from 1 to 3 hours between patients.
Busulfan
Because of its insolubility in aqueous solutions, busulfan has previously been available only as an oral preparation. However, an intravenous preparation has been recently made available251 but has not been extensively used, so that most of the published pharmacokinetic information is after oral administration. For myeloablative therapy prior to bone marrow transplantation, busulfan is widely used at a dose of 1 mg/kg every 6 hours for 4 days. After a 1 mg/kg dose in adults and older children, there is a considerable variation in bioavailability, with peak plasma levels of 1 to 10 micromolar and elimination half-times between 1 and 7 hours.252–254 The AUC after a single dose in adults and older children varies between 10 and 80 micromolar hours. However, in young children (age 1–3), the peak plasma concentrations are less (1–5 micromolar), the mean elimination time about 40% faster, and the AUC consistently less at 6 to 17 micromolar hours.255 Grochow and colleagues252 have demonstrated that AUCs of busulfan greater than one standard deviation from the mean values for all patients are associated with a very high risk of veno-occlusive disease of the liver (Fig. 48.17). It has now been demonstrated that pharmacokinetic guided adjustment of the busulfan dose can reduce the incidence and severity of this toxicity.256,257
Figure 48.17
Structures of platinum antitumor agents.
Toxicities
The characteristic toxicities of the alkylating agents are hematopoietic, gastrointestinal, gonadal, and CNS toxicity. However, each of the agents has a characteristic set of toxicities, determined by the reactivity, metabolism, and distribution of the agent, and the clinician should be aware of these idiosyncrasies of the agents.
Hematopoietic Toxicity
In general, the clinical dose-limiting toxicity for alkylating agents is hematopoietic toxicity, particularly suppression of granulocytes and platelets. The nadir of granulocyte depression after alkylating agents is usually 8 to 16 days, and the granulocytes usually return to normal within 20 days after a single dose of the agent.258 Cyclophosphamide and ifosfamide are less hematopoietically toxic than other alkylating agents258,259; granulocyte levels return to normal more rapidly, platelets are affected less, and repeated doses of cyclophosphamide and ifosfamide do not produce cumulative damage and progressive deterioration of the hematopoietic elements. The reduced hematopoietic toxicity of cyclophosphamide and ifosfamide is due to the presence of aldehyde dehydrogenase in the hematopoietic stem cells and the early megakaryocytes, as discussed earlier. In contrast, the nitrosoureas produce severe hematopoietic toxicity, with a delayed onset and nadirs of granulocytes and platelets occurring as late as 45 days.75,260 Busulfan also produces severe hematopoietic depression, with a selectivity for early myeloid precursors.67,68 The variations in the cellular patterns and time courses of hematopoietic suppression after the administration of different alkylating agents indicate that the individual agents have selectivity for different hematopoietic precursor cells (Fig. 48.18).

Figure 48.18
Aquation of platinum compounds and reaction with nucleophiles.
Peptide growth factors, such as granulocyte-macrophage colony-stimulating factor (sargramostim, GM-CSF) and granulocyte colony-stimulating factor (filgrastim, G-CSF), which stimulate the differentiation and proliferation of hematopoietic precursors,261 are now used clinically. The degree and duration of granulocyte depression after antitumor drug administration can be reduced by the concomitant use of these growth factors.262–264 Currently, growth factors that may stimulate the proliferation and restoration of megakaryocytes and platelets are under investigation.265,266 The use of these factors with the alkylating agents has been particularly attractive because of the steep dose-response curve of the alkylating agents and because, with several alkylating agents, a considerable increase in dose may be administered before another dose-limiting toxicity is reached. For these same reasons, combinations of alkylating agents have been used extensively in association with allogeneic and autologous bone marrow transplantation.71,267
Gastrointestinal Toxicity
Damage to the gastrointestinal tract is a toxicity that frequently occurs with high-dose regimens. Mucositis, stomatitis, esophagitis, and diarrhea occur with high doses of alkylating agents and in particular after high doses of melphalan and thiotepa or combinations of alkylating agents including melphalan or thiotepa.268–270 Significant mucositis is unusual even after very high doses of cyclophosphamide or ifosfamide. This lack of gastrointestinal toxicity is probably due to the presence of the enzyme aldehyde dehydrogenase in the epithelial cells of the gastrointestinal tract.13
Nausea and vomiting are frequent side effects of alkylating agents. Although these side effects are not usually life threatening, they are major discomforts to patients and may result in the delay or discontinuation of therapy. The nausea and vomiting are, at least in part, mediated through the CNS and are not due to direct gastrointestinal toxicity.271,272 These effects are variable between patients in that some people tolerate high doses of these drugs without nausea and vomiting, whereas other patients are incapacitated by even low doses of alkylating agents. The frequency of nausea and vomiting does increase as the dose of alkylating agents is increased. Therefore, it is important, especially with the use of increasing doses of alkylating agents, to provide the patient with adequate antiemetic medication. Such medications include phenothiazines, other antiemetics, acute doses of corticosteroids, and, more recently, antiserotonin agents.273–275
Veno-Occlusive Disease of the Liver
This syndrome is characterized clinically by hepatomegaly, right upper quadrant pain, jaundice, ascites, and a high mortality rate from hepatic failure. Pathologically, the syndrome is associated with subendothelial thickening and narrowing of the hepatic venule lumen.276 This complication has been seen in about 25% of patients receiving high-dose cyclophosphamide and busulfan (see Fig. 48.17) or cyclophosphamide and total body irradiation prior to allogeneic or autologous bone marrow transplantation for leukemia or lymphoma,276 and has also been seen after other high-dose alkylating agent therapy.277,278 Liver transplantation has been used for the treatment of veno-occlusive disease in patients after bone marrow transplantation.279,280
Gonadal Damage
A serious toxicity of the alkylating agents is gonadal damage. The characteristic lesion in men, depletion of testicular germ cells with preservation of Sertoli cells, was first described in 1948 in patients treated with mechlorethamine.281 This lesion has subsequently been observed with other alkylating agents282 and frequently results in aspermia or oligospermia in men treated with drug combinations including alkylating agents.283 However, spermatogenesis and fertility may return after several years.284,285
Amenorrhea, associated with disappearance of mature and primordial ovarian follicles, is seen in women treated with alkylating agents.70,286,287 The frequency of amenorrhea increases with the age of the woman and is more likely to be irreversible in older women.288
Pulmonary Damage
Pulmonary damage in the form of interstitial pneumonitis and fibrosis has been associated with almost all of the alkylating antitumor drugs. Although the exact mechanism of the pulmonary toxicity is not known, it is presumably due to direct toxicity of the alkylating agents to pulmonary epithelial cells. The typical presentation of this toxicity is the onset of a nonproductive cough and dyspnea, which may progress to tachypnea and cyanosis and even to severe pulmonary insufficiency and death. This complication was first described in association with busulfan therapy,289 but subsequently it has been described after cyclophosphamide,290,291 nitrosoureas,292,293 melphalan,294 chlorambucil,295 and mitomycin C.296 A significant incidence of pulmonary toxicity has been reported in patients receiving high doses of cyclophosphamide, cisplatin, and BCNU.297
Hemorrhagic Cystitis
The oxazophosphorines, cyclophosphamide and ifosfamide, produce bladder toxicity, which is not seen with other alkylating agents. This toxicity is a hemorrhagic cystitis, which may progress to massive hemorrhage.298,299 The toxicity has been demonstrated to be due to metabolites of these drugs, which are excreted into the urine. The metabolite principally responsible for this toxicity is acrolein,300 although phosphoramide mustard and chloracetaldehyde may contribute to the effect. Hemorrhagic cystitis is seen more commonly after ifosfamide therapy than cyclophosphamide, partly because higher doses of this agent are used (see above). Renal tubular damage has also been seen after ifosfamide, including a Fanconi-type syndrome with azotemia, elevated serum creatinine, and enzymuria.301
The systemic administration of thiols can prevent or ameliorate the bladder damage from cyclophosphamide and ifosfamide because the thiols conjugate the aldehyde functions of acrolein and chloracetaldehyde. The most widely used compound to prevent oxazophosphorine bladder toxicity is the sodium salt of 2-mercaptoethane sulfonate (MESNA).302 MESNA is usually administered to all patients receiving ifosfamide and to patients who are receiving high-dose cyclophosphamide. Subclinical renal toxicity has been observed in children receiving ifosfamide,16,303 despite MESNA administration, so that administration of MESNA does not eliminate the need for adequate hydration and careful observation of the patient.
Antidiuresis
An antidiuretic effect is commonly seen in patients receiving doses of cyclophosphamide of 50 mg/kg or greater.304,305 This syndrome is characterized by a decrease in urine output 6 to 8 hours after drug administration, weight gain, a marked increase in urine osmolality, and a decrease in serum osmolality and sodium concentration. Pericardial and pleural effusions may be seen, and seizures due to hyponatremia have occurred after cyclophosphamide therapy,306 especially if low-sodium replacement fluids have been administered. This antidiuretic syndrome appears to be due to an effect of cyclophosphamide metabolites on the distal renal tubule and is self-limited, with the excess fluid excreted over a period of about 12 hours. Administration of furosemide will promote free water clearance and ameliorate the syndrome.307
Renal Toxicity
Renal toxicity has proven to be a serious toxicity of the nitrosoureas.308,309 This effect is dose-related and may produce severe renal failure and death after administration of more than 1,200 mg of BCNU. Elevation of serum creatinine and other clinical evidence of renal toxicity may not be seen until after the completion of therapy. The histology of the kidneys in patients with renal nitrosourea damage is similar to that in radiation nephritis. A case of acute renal failure after melphalan therapy has been reported.310
Alopecia
Although the association between an alkylating agent and alopecia was first described with busulfan therapy,311 this toxicity has been predominantly associated with cyclophosphamide and ifosfamide therapy. The alopecia produced by these agents may be quite severe, especially if the agent is given in combination with vincristine or doxorubicin. Regrowth of the hair occurs after cessation of therapy and may be associated with a change in the texture and color of the hair.312 The structure-function studies of Feil and Lamoureaux313 suggest that this toxicity is due to the entry of lipophilic metabolites into the hair follicles. This suggestion is consistent with the fact that busulfan, vincristine, and adriamycin are all lipophilic molecules.
Allergic and Hypersensitivity Reactions
Since the alkylating agents react with many biologic molecules, it is not surprising that they would serve as haptenes and produce allergic reactions.314–316 The most frequent reactions that have been reported have been cutaneous hypersensitivities. Anaphylactic reactions are rare, but they have occurred.317 Patterns of cross-reactivity have not been carefully defined, but cross-reactivity between agents of similar structure, such as the nitrogen mustards, have been described.316,318
Cardiotoxicity
The nonhematologic dose-limiting toxicity of cyclophosphamide is cardiac toxicity.319–321 The fulminant syndrome has been seen most frequently in patients receiving a total dose of cyclophosphamide greater than 200 mg/kg preparatory to bone marrow transplantation. The clinical course of the syndrome consists of the rapid onset of severe heart failure, which is fatal within 10 to 14 days. The hearts of such patients are dilated, with patchy transmural hemorrhage and pericardial effusion. The microscopic findings consist of interstitial hemorrhage and edema, myocardial necrosis and vacuolar changes, and specific changes in the intramural small coronary vessels.320 Decreased electrocardiographic voltage and a transient increase in heart size is seen in high-dose cyclophosphamide patients without clinical symptoms, and the characteristic pathologic findings are present in such patients who die of other causes. Cardiotoxicity and cardiomegaly have been seen in patients receiving lower doses of cyclophosphamide in combination with other alkylating agents.322 Age greater than 50 and previous adriamycin exposure appear to increase the risk of cyclophosphamide cardiotoxicity.321
Neurotoxicity
In preclinical studies of alkylating agents, convulsions have often been seen.323 At the usual clinical doses of these agents, frank neurotoxicity is not usually seen but drowsiness and alterations of consciousness can be seen.324 With the increasing use of higher doses of alkylating agents and combinations of alkylating agents, more clinical neurotoxicity is being seen.325 At BCNU doses of 1,200 mg/m2, severe CNS toxicity has been seen,326 and the intracarotid administration of BCNU has produced severe eye pain and blindness.327 High-dose busulfan therapy produces seizures, and anticonvulsants are often used prophylactically in these patients.328
Teratogenecity
Studies carried out in vivo and in embryo cultures have demonstrated that virtually all of the alkylating agents are teratogenic.329,330 The teratogenic effect is probably due to cytotoxic effects on the embryo by the same mechanisms by which the compounds are toxic to tumor cells.331–334 The available clinical information indicates that there is a definite risk of a malformed infant if the mother is treated with an alkylating agent during the first trimester of pregnancy.335–337 In a review of the literature, Nicholson338 found that of 25 women who had received alkylating agents during the first trimester of pregnancy there were four fetal malformations. However, the administration of alkylation agents during the second and third trimesters is not associated with an increased risk of fetal malformation.338–340
Carcinogenesis
Since the initial reports of acute leukemia occurring in patients treated with alkylating agents,341–344 it has become increasingly obvious that this type of oncogenesis is a significant complication of alkylating agent therapy. Several studies have indicated that the rate of acute leukemia after alkylating agent therapy may be 10% or higher in certain groups of patients.345–347 Procarbazine and other methylating agents appear to be the most potent oncogenic agents,348 and melphalan appears to produce a higher rate of acute leukemia than cyclophosphamide.349 The lesser leukemogenic potential of cyclophosphamide may well be related to the hematopoietic stem cell sparing effect of this agent.13 An increased rate of solid tumors is also seen in patients treated with alkylating agents.350,351 Although sufficient data are not yet available to be certain, it appears that high-dose alkylating agent therapy administered in intermittent pulses over a relatively short period of time is less oncogenic than prolonged alkylating agent therapy.
Immunosuppression
The immunosuppressive effect of alkylating agents was first described by Hektoen and Corper352 for sulfur mustard. Cyclophosphamide is particularly immunosuppressive353 and is used for the treatment of autoimmune diseases,354–356 Cyclophosphamide is also used in preparative regimens for allogeneic transplantation because of its immunoablative activity.357 Low doses of cyclophosphamide and melphalan can enhance the immune response by selectively inhibiting the immune suppressor cells.358–360 Because of this effect, moderate doses of cyclophosphamide have been used in conjunction with immunotherapy and biologic response modifiers, such as interleukin-2.361,362
The clinical significance of the immunosuppression produced by alkylating agents in their role as antitumor agents is not certain. The two major concerns are susceptibility to infection in the immunosuppressed host and the potential interference with a host immune response to the tumor. The available evidence indicates that most intermittent antitumor regimens do not produce a profound or prolonged immunosuppression.363
Platinum Antitumor Compounds
The platinum antitumor agents are complexes of platinum with ligands that can be displaced by nucleophilic (electron-rich) atoms to form strong bonds with covalent characteristics. Thus, like the alkylating agents, the platinum agents form strong chemical bonds with thiol sulfurs and amino nitrogens in proteins and nucleic acids.
The first platinum antitumor compound was discovered by Rosenberg and colleagues364,365 while studying the effects of electric current on bacterial growth. The growth inhibition observed was found to be caused by a platinum complex of ammonia and chloride, which was produced in the medium from the platinum electrode. These investigators found several such compounds to have antitumor activity against murine tumors in vivo.365 The most active of these compounds was the one now known as cisplatin (Fig. 48.19).
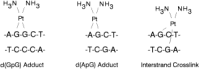
Figure 48.19
Platinum-DNA adducts.
Cisplatin went into clinical trials in the early 1970s 366–368 and was found to have significant antitumor activity against testicular cancer, lymphoma, squamous cell carcinoma of the head and neck, ovarian cancer, and bladder cancer. Because of its significant therapeutic effect in these tumors and activity against a number of other solid tumors, it became the most frequently used antitumor agent. Because of the renal and neurotoxicities of cisplatin, there were intensive efforts to devise analogues with fewer of these toxicities. This work led to the development of carboplatin, which produces primarily hematopoietic toxicity and appears to have an antitumor effect similar to cisplatin 369–373 against the tumors against which it has been used. A number of other platinum compounds are currently under investigation and are discussed below.
Chemistry
The platinum compounds that are active antitumor agents can have either four or six ligands (see Fig. 48.19), with a square planar or hexahedral configuration, respectively. Those with four ligands have an oxidation state of +2, and those with six ligands an oxidation state of +4. The chloride ligands of cisplatin and the other complexes with the 12 oxidation state can be exchanged for nucleophilic atoms in the biologic milieu, including the nitrogens of the DNA bases. The chloride ligands of the +4 compounds are much less reactive than those of the +2 compounds,374 and it is likely that the +4 compounds are reduced in vivo to produce the reactive +2 complexes.375–377 The ligand substitution reactions of the square planar complexes occur with retention of the configuration of the platinum complex.378 Since the trans-platinum compounds are essentially inactive as antitumor compounds, the ability of the cis compounds to form certain stereo-specific cross-links probably accounts for their antitumor activity.
In some cis-platinum compounds in clinical use, the chloride leaving ligands are replaced with carboxyl ester groups, as in carboplatin and oxaliplatin (see Fig. 48.19). These ligands are less readily displaced; thus, these compounds require higher concentrations for cytotoxicity. The decreased renal and neurologic toxicity of these compounds is also probably due to the fact that they are less chemically reactive than cisplatin. Substitutions on the amino groups alter the lipophilicity and distribution of the agent.
Cellular and Molecular Pharmacology
Although the chloride and carboxyester ligands can probably be directly displaced by biologic atoms, it is likely that, in the biologic milieu, the chloride or carboxy ligands are displaced by water molecules to form the aquo ligand, which is a better leaving group than the chloride or carboxy groups.378 The high chloride content of the extracellular fluid maintains the platinum compounds in the chloride and less reactive form. However, in the lower chloride content of the cell, the more reactive aquo species is formed. The loss of a proton produces the hydroxy ligand, which is unreactive.379 The proposed aquation pathway for cisplatin is shown in Figure 48.20. The platinum compounds react with many biologic molecules, but there is considerable evidence that these compounds, like the bifunctional alkylating agents, exert their cytotoxic effect by reacting with DNA and interfering with DNA replication and cell division. Roberts and Pera380 demonstrated that the amount of platinum bound to DNA was directly related to the degree of toxicity of platinum compounds. Zwelling and colleagues381 demonstrated that the degree of DNA interstrand cross-linking in vitro and in vivo was directly related to the degree of cytotoxicity in rodent tumor cells.
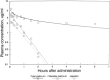
Figure 48.20
Clinical pharmacokinetics of cisplatin after single injection of 100 mg/m2. (Adapted from Patton et al.
The cis-platinum compounds, like the alkylating agents,382–384 react with nitrogen atoms of DNA and preferentially react with the N-7 atom of deoxyguanylic acid. Specific adducts of Pt compounds with DNA have now been characterized and studied.380 The consensus of the studies is that the most frequent adducts are dGpdG and dApdG, which result from the cisplatinum complex binding to adjacent deoxyguanylates or an adjacent deoxyadenylate and deoxyguanylate in a strand of DNA to produce an intrastrand cross-link in both situations. A less common lesion is one that results from binding of the platinum atom to the N-7 of a deoxyguanylate in one strand of DNA and to the N-7 atom of a deoxyguanylate in the complementary strand of DNA, thereby producing an interstrand cross-link. Repair of these lesions does occur, and the cytotoxicity to the cell is probably determined by the resultant formation and repair of the lesions.385 As mentioned above, a close correlation between interstrand DNA cross-linking has been demonstrated, but equally precise methods for quantifying intrastrand cross-links in whole cells after drug exposure are not available. Thus, intrastrand DNA cross-links might correlate equally well or better with cytotoxicity. The DNA adducts formed by Pt compounds other than cisplatin have been less well studied but appear to be similar to those formed by cisplatin.386–388
Although there is considerable evidence that the formation of DNA adducts is responsible for the cytotoxicity of the platinum antitumor agents, the mechanisms through which the cytotoxic effects are mediated are not well understood. Evidence has been presented that the platinum adducts inhibit replication.389,390 Heiger-Bernays and colleagues391 have demonstrated that as few as two platinum adducts per genome were sufficient for inhibition of DNA replication by cisplatin. Sorenson and Eastman392 found that cytotoxicity with cisplatin was correlated with the duration of arrest in the G2 phase of the cell cycle and postulated that the G2 arrest was due to the inability of the cells to transcribe the Pt-damaged DNA and produce the mRNA essential for mitosis.
In 1996, Drummond and colleagues393 demonstrated that ovarian tumor cells resistant to cisplatin were deficient in the MutL alpha MLHI subunit and suggested that the mismatch repair system recognized the cisplatin cross-link and played a role in the cytotoxicity of cisplatin. Similar findings were reported by others,394 and Mello and colleagues395 postulated that the mismatch repair protein hMSH2 played an active role in mediating cisplatin cytotoxicity. Vaisman and colleagues have suggested that mismatch repair defects result in increased replicative bypass of cisplatin adducts.396 It has also been established that transplatin lesions, and those produced by oxaliplatin, tetraplatin, and JM-216, are not recognized by the mismatch repair system,397 and that these agents produce cytotoxicity in mismatch repair deficient cells. The latter three agents produce a cross-link containing the bulky cyclohexylamino group (see Fig. 48.19). Takahara and colleagues And Gelesco and Lippard have now reported both the crystal structure398 and the NMR solution structure399 of the cisplatinum d(GpG) cross-link. This type of structural understanding should increase the interpretation of the functional effects of the platinum cross-links.
Mechanisms of Cellular Resistance to Platinum Agents
A number of mechanisms of cellular resistance to platinum compounds have been described. These mechanisms include decreased uptake of the platinum compound into resistant cells, inactivation of the drug by cellular thiol compounds, enhanced repair of the platinum-related DNA damage, and the absence of mismatch repair, as described above.
Decreased cellular uptake of cisplatin by cells resistant to the compound has been described by a number of investigators.400–404 The uptake of cisplatin into cells is linear for over an hour and does not appear to be an active transport process, although it is partially inhibited by metabolic inhibitors.400 There has also been a report of increased efflux of cisplatin in a resistant cell line.405 Mann and colleagues406 could not demonstrate changes in the physical properties of the cell membrane of cells resistant to cisplatin. Thus, although decreased cellular accumulation of the platinum compounds appears to be one type of cellular resistance, the mechanism of this type of resistance remains undefined and may be related to altered binding of the agents to cellular proteins, rather than alteration of passage through the cell membrane.407
A number of investigators have demonstrated that both rodent and human tumor cells that are selected in vitro or in vivo by exposure to the platinum antitumor compounds frequently demonstrate elevated glutathione levels in association with resistance to these drugs.408–413 Tumor cell lines derived from patients resistant to therapy with cisplatin have also been found to have elevated glutathione levels.414,415
Further evidence that glutathione is involved in resistance to platinum compounds can be inferred from the fact that several investigators have shown that tumor cells can be sensitized to the platinum agents by depletion of cellular glutathione by treatment with buthionine sulfoximine, an inhibitor of glutathione synthesis.413–418
The mechanism(s) through which glutathione-associated resistance is mediated have not been definitively elucidated. Andrews and colleagues416 demonstrated that cisplatin binds to glutathione, and Dedan and Borch419 have studied the reaction rates of cisplatin with various thiols, including glutathione, and characterized a reaction product in which two glutathiones appeared to be bound to each platinum through the cysteine residues of the glutathiones. The thiol platinum ligand is very stable and thus will not react further. Eastman410 has presented evidence that glutathione may react with monofunctional adducts on DNA to quench the second reactive ligand and prevent cross-link formation. Resistance to cisplatin has also been associated with elevation of glutathione transferase enzyme activity, increased levels of the pi (acidic) isozyme of the protein, and increased levels of the mRNA for the pi isozyme.420–422 However, the catalysis of the conjugation of glutathione with platinum agents by this enzyme has not been characterized.
Cellular resistance to platinum agents has also been associated with another sulfhydryl-containing protein, metallothionein. Several investigators have found that tumor cells exposed to heavy metals, such as cadmium, develop resistance to cisplatin, which is associated with increased cellular levels of metallothionein.423–425 In one report, transfection of cells with the metallothionein gene resulted in increased metallothionein levels and resistance of the cells to cisplatin, melphalan, and chlorambucil.425 Naganuma and colleagues426 have reported that administration of bismuth subnitrate to mice produced increased levels of metallothionein in the kidneys and resulted in protection of the mice from the renal and gastrointestinal toxicity of cisplatin but did not affect the response of transplanted tumors to cisplatin in the mice. Cisplatin binds to metallothionein in Ehrlich ascites tumor cells427 and in the liver and kidney of rats,407,428 and the systemic administration of cisplatin or its hydrolyzed product can induce metallothionein in the liver and kidney.429 These findings indicate that metallothionein can protect both tumor and normal cells from cisplatin, although the binding of the drug to this protein has not been characterized.
As with the alkylating agents, there is extensive evidence that enhanced DNA repair can be responsible for resistance to the platinum compounds. Van Den Berg and Roberts430 first reported that caffeine, a known inhibitor of DNA repair, potentiated cytotoxicity and chromosomal damage in mammalian cells, and shortly thereafter Fravel and Roberts demonstrated that excision repair of cisplatin-damaged DNA does occur in treated cells.431 Many subsequent studies have demonstrated that cells deficient in DNA repair, such as those from patients with xeroderma pigmentosum or Fanconi’s anemia, are very sensitive to cisplatin.432–436
Agents that are known to inhibit the activity of enzymes involved in the repair of DNA, such as aphidocolin and novobiocin, have been shown to sensitize cells to cisplatin and to reverse the resistance of repair-resistant cell lines.437–440 The antitumor agents hydroxyurea and cytosine arabinoside, which inhibit DNA repair synthesis, both produce a synergistic cytotoxic effect with cisplatin.441,442
Studies by Becket and colleagues And Husain and colleagues have supported the above indications that platinum adducts in DNA are repaired by an excision repair mechanism,443,444 and these investigators445 have now reported that the nucleotide excision repair system can remove DNA adducts produced from cisplatin, oxaliplatin, and JM 216 (Bis-aceto-ammine-dichloro-cyclohexylamine-platinum IV). These findings are consistent with studies by Dabholkar and colleagues and Li and colleagues demonstrating that ERCC-1 is involved in platinum repair.446,447
It has also been shown that platinum interstrand DNA cross-links are removed more rapidly in cisplatin-resistant cells448 and that very sensitive tumor cells may have a decreased ability to remove DNA interstrand cross-links.449 A protein, XPE-BF (xeroderma pigmentosum complementation group E binding factor), which binds to Pt-damaged DNA450–452 and may mark it for repair, has been identified. A series of proteins, the HMG domain proteins, which bind to Pt intrastrand cross-links, produce bending of the DNA, and may inhibit the repair of these lesions, has also been described.453,454 It has also been found that cisplatin-resistant cells can have elevated thymidylate synthase activity and be cross-resistant to 5-fluorourocil (5FU)455 and that c-fos may play a role in the cellular response to Pt agent damage by mediating DNA repair pathways.456–458
Although it is clear that each of these mechanisms can be associated with the resistance of tumor and normal cells to the platinum agents, the relative roles of these mechanisms in the resistance of tumors to treatment in patients have not been established. Such studies and attempts to overcome resistance with BSO and inhibitors of DNA repair are currently in progress.
Clinical Pharmacology
Analogues in Clinical Use
Cisplatin and carboplatin are licensed in the United States and internationally and are used extensively. Since the primary toxicity of carboplatin is hematopoietic, it has replaced cisplatin for use in many patients and is being used particularly in situations where nonhematopoietic toxicity should be avoided, such as high-dose treatment with bone marrow support459,460 or with hematopoietic stimulatory factors. There is no evidence for cross-resistance between these two agents. Iproplatin has been evaluated in phase II trials but was found to be no more or less effective than carboplatin and produced more hematopoietic and gastrointestinal toxicity.461–465 Tetraplatin (ormaplatin) produced severe neurotoxicity in initial clinical trials466 but is still being evaluated in phase I trials.467 Oxaliplatin is similar to tetraplatin in its preclinical toxicity.468 However, this compound has shown promising activity in gastrointestinal tumors, especially in combination with 5-FU and leucovorin.469–471 Oxaliplatin has also demonstrated significant activity in patients with ovarian cancer who have previously received cisplatin.472, 473Oxaliplatin demonstrated a modest effect (15% PR) in advanced, cisplatin-resistant non-small-cell lung patients.474
A lipid-soluble platinum compound, JM216,475 which can be administered orally, is now being evaluated clinically.476–478 The discovery that oxaliplatin and JM216 are active against tumors lacking mismatch repair (see above) has stimulated interest in these and related compounds, and some of the new platinum analogues may not be totally cross-resistant with cisplatin due to differences in either cellular uptake402 or cellular detoxification.479
Pharmacokinetics
Platinum antitumor compounds have been measured in human plasma and other human tissues as total platinum, as ultrafilterable platinum, and as the specific parent compounds. Total platinum can be measured by using compounds containing the radioactive 193Pt or 195Pt isotopes480,481 by trapping the platinum with an ultraviolet absorbing ligand, such as diethyldithiocarbamate,482 or by flameless atomic absorption spectroscopy.482,483 Ultrafiltration of plasma and other biologic fluids separates the free platinum compounds from those bound to protein. The protein-bound species are biologically inactive and essentially irreversibly bound to the protein.484 Both cisplatin and carboplatin have been measured specifically by separation from other species on HPLC columns and detection by electrochemical detection or by collecting fractions and quantifying the total platinum in each fraction.485,486 The cisplatinum concentration has been found to be consistently between 60 and 80% of the ultrafilterable platinum and to follow the same kinetics as the ultrafilterable platinum .487,485 Carboplatinum represents a higher percentage of the ultrafilterable platinum and follows kinetics similar to the ultrafilterable platinum. Because of the sensitivity, accuracy, and convenience of the method, flameless atomic absorption spectroscopy is the most common technique used to measure the platinum agents. Furthermore, since measuring filterable species appears to measure the reactive compounds and to approximate closely the measurement of the parent compounds, measurement of ultrafilterable platinum is most commonly used in pharmacokinetic studies.
In pharmacokinetic studies after cisplatin administration, total platinum in the plasma follows a triphasic pattern, with the first phase t 1/2 about 30 minutes, the second phase t1/2 about 60 minutes, and the third phase t1/2 greater than 24 hours.487,488 Measurements of the ultrafilterable platinum indicate that the initial, more rapid clearance phases are due to the renal clearance of filterable platinum, the majority of which is the parent compound.488 Carboplatin exhibits similar pharmacokinetics, except that the initial half-lives are somewhat longer, less of the total platinum is protein bound, and a greater percentage of the agent is excreted by the kidneys.486,489 The pharmacokinetics of total and filterable platinum after iproplatin administration appears to be similar to those of carboplatin.490 Decreased creatinine clearance results in higher plasma levels of both cisplatin and carboplatin and potentially greater toxicity.
After bolus administration of 100 mg/m2 of cisplatin, initial peak plasma concentrations of 3 to >5 microgram/ml are achieved,485 with this value decreasing to less than 0.2 microgram/ml at 2 hours. After the usual clinical dose of about 300 mg/m2 of carboplatin, peak plasma levels of about 30 microgram/ml are reached, declining to about 5 microgram/ml at 2 hours.486,489
In typical clinical use, usually in combination with other agents, the platinum antitumor agents are given intravenously, either as a single dose or daily for several days, with repeat courses at 3 to 4 weeks. The agents are given as an infusion over several hours rather than as a bolus dose and, especially with very high doses, may be given as 24-hour or longer infusions. Because of the close relationships between plasma AUC of carboplatin and renal function and between AUC of carboplatin and toxicity, dosing algorithms based on renal function have been established and are now widely used in the dosing of carboplatin.491–493
Cisplatin and carboplatin have also been administered regionally. There has been considerable experience with the intraperitoneal route, particularly in the treatment of ovarian cancer.494–496 Very high intraperitoneal concentrations can be obtained, and systemic toxicities can be reduced by the concomitant systemic administration of thiosulfate.497,498 Cisplatin has also been administered intra-arterially for the treatment of tumors in the extremities,499–502 brain tumors,503–505 carcinoma of the head and neck,506,507 carcinoma of the liver,508 and carcinoma of the bladder.509,510 Intravesicular instillation of cisplatin has been used for the treatment of superficial cancers of the bladder.511–513 Cisplatin has also been instilled into the pericardial sac for the treatment of malignant pericardial effusions.514,515
Toxicities
Renal
The most serious, and usually dose-limiting, toxicity of cisplatin is renal.516,517 This toxicity is manifested clinically by elevated BUN and creatinine, is cumulative with continued cisplatin exposure, and is potentiated by other nephrotoxins.518 Decreases in serum electrolytes have been associated with platinum renal toxicity, including symptomatic hypomagnesemia.519 Although the toxicity may remain subclinical, or the renal function return to normal, significant pathologic damage appears to persist.520 The pathology of the renal damage is characterized by focal acute tubular necrosis, dilatation of convoluted tubules, thickened tubular basement membranes, formation of casts, and epithelial atypia of the collecting ducts.520,521 High fluid intake with forced diuresis522,523 can reduce the incidence and severity of the renal toxicity. Systemic administration of thiols can reduce renal toxicity of cisplatin in animal models, and in a clinical trial, systemic diethyldithiodicarbamate appeared to reduce nephrotoxicity without affecting ototoxicity or myelosuppression.524 The nephrotoxicity of the second-generation platinum complexes, such as carboplatin and iproplatin, is markedly less than that of cisplatin.
Ototoxicity
Ototoxicity has been a significant problem with cisplatin. This toxicity is characterized by tinnitus and hearing loss.366,367,525 The hearing loss is usually in the high-frequency range, 4,000 to 8,000 Hz, but may occur in the lower ranges, which include the speech frequencies.525,526 Since the higher frequencies are usually involved, the hearing loss may not be symptomatic. Vestibular toxicity does not usually occur but can be seen.527,528 The ototoxicity of cisplatin is dose related and is usually cumulative with subsequent courses of the agent.529,530 Radiation prior to or simultaneous with the cisplatin administration enhances the toxicity,531,532 but this additive effect may be less if the cisplatin precedes the radiation.526
The pathologic findings associated with ototoxicity, in both experimental animals and patients, are selective damage to the outer hair cells of the cochlea and lesions in the organ of Corti, the spiral ganglion and cochlear nerve, and the stria vascularis.533–536 In studies of organ cultures of the cochlear structures, the hair cells are very sensitive to very low concentrations of cisplatin.537 Vestibular toxicity is associated with degeneration of the maculae and cristae.528
Neurotoxicity
The neurotoxicity seen with the administration of cisplatin consists principally of peripheral neuropathy involving both the upper and lower extremities, with paresthesias, weakness, tremors, and loss of taste.538 Seizures and leucoencephalopathy have also been described.539–542 The neurotoxicity may be persistent543 and may progress after cessation of cisplatin therapy.542 The quantitative determination of vibratory perception threshold has been reported to correlate with cisplatin neurotoxicity.544
Particularly severe neurotoxicity has been reported after intra-arterial infusions of cisplatin, with cranial nerve paralysis occurring after intra-arterial infusions for head and neck cancer541,542 and severe peripheral neuropathy after lower limb perfusion.545 In experimental animals, severe CNS toxicity was seen when compounds that open the blood-brain barrier were administered prior to systemic cisplatin treatment, and intracarotid cisplatin produced damage to the blood-brain barrier and severe neurotoxicity.546 However, severe neurotoxicity was not seen in patients treated with intracarotid cisplatin for primary brain tumors.547 The neurotoxicity of ifosfamide has been reported to be enhanced by prior treatment with cisplatin.548
Since various pharmacologic maneuvers have been able to control or reduce the nephrotoxicity and severe nausea and vomiting produced by cisplatin, neurotoxicity has become the dose-limiting toxicity of cisplatin.549 An interesting observation is that treatment of animals with an ACTH analogue will prevent neurotoxicity from cisplatin and will facilitate the recovery of established neurotoxicity550,551 but will not interfere with the antitumor effect of the agent. In a randomized, placebo-controlled clinical trial, this compound appeared to prevent or ameliorate the neurotoxicity of cisplatin.551 Neither carboplatin or iproplatin appear to produce significant neurotoxicity with the doses used thus far with autologous bone marrow transfusion.552–554
Gastrointestinal Toxicity
Severe nausea and vomiting have been a significant problem with cisplatin, occurring in almost all patients receiving the drug.517,555 The cause of this toxicity is not firmly established. Work in animal models indicates that abdominal visceral innervation and 5-hydroxytryptamine receptors on visceral afferent nerves play a role in mediating this toxicity,556 but there is also evidence that the chemoreceptor trigger zone in the medulla plays a role.557,558 The use of a dopamine antagonist, metoclopramide, prior to and during cisplatin administration has been effective in controlling this toxicity,559,560 and the steroids dexamethasone or methylprednisolone alone or in combination with metoclopramide have also been useful.561–563 More recently, antiserotonin analogues such as ondansetron and granisetron have proven highly effective in controlling nausea and vomiting after platinum administration. The gastrointestinal toxicities of carboplatin and iproplatin are much less than those of cisplatin.564–566
Immune Effects
In contrast to the alkylating agents, many of which are significantly immunosuppressive, cisplatin appears to have no immunosuppressive effect at the usual clinical doses and may even augment immune function at these doses.567 Monocyte-mediated cytotoxicity was found to be increased in ovarian cancer patients after cisplatin treatment,568 and OKT81 cytotoxic cells were increased in patients after cisplatin therapy.569
References
- 1.
- Adair C P J, Bogg H J. Experimental and clinical studies on the treatment of cancer by dichloroethylsulfide (mustard gas). Ann Surg. 1931;93:190. [PMC free article: PMC1398743] [PubMed: 17866462]
- 2.
- Goodman L S, Wintrobe M M, Dameshek W, Goodman J J. et al. Use of methyl-bis(beta-chlorethyl)amine hydrochloride for Hodgkins disease, lymphosarcoma, leukemia. JAMA. 1946;132:263. [PubMed: 20997191]
- 3.
- Jacobson L P, Spurr C L, Barron E S G. et al. Studies on the effect of methyl-bis(beta-chloroethyl)amine hydrochloride on neoplastic diseases and allied disorders of the hematopoietic system. JAMA. 1946;132:263. [PubMed: 20997209]
- 4.
- Rhoads C P. Nitrogen mustards in treatment of neoplastic disease. JAMA. 1946;131:656. [PubMed: 20984885]
- 5.
- Colvin M, Chabner BA. Alkylating Agents in Cancer Chemotherapy: Principles and Practice. Edited by BA Chabner, JM Collins. Philadelphia: Lippincott, 1990,pp 276–313.
- 6.
- Fenselau C, Kan M N, Rao S S. et al. Identification of aldophosphamide as a metabolite of cyclophosphamide in vitro and in vivo in humans. Cancer Res. 1977;37:2538–2543. [PubMed: 872080]
- 7.
- Zon G, Ludeman S M, Brandt J A. et al. NMR spectroscopic studies of intermediary metabolites of cyclophosphamide. A comprehensive kinetic analysis of the interconversion of cis- and trans-4-hydroxycyclophosphamide with aldophosphamide and the concomitant partitioning of aldophosphamide between irreversible fragmentation and reversible conjugation pathways. J Med Chem. 1984;27:466. [PubMed: 6708049]
- 8.
- Shulman-Roskes E M, Noe D A, Gamcsik M P. et al. The partitioning of phosphoramide mustard and its aziridinium ions among alkylation and P-N bond hydrolysis reactions. J Med Chem. 1998;41:515–529. [PubMed: 9484502]
- 9.
- Alarcon R A, Meienhofer J. Formation of the cytotoxic aldehyde acrolein during the in vitro degradation of cyclophosphamide. Nature New Biol. 1971;233:250–252. [PubMed: 20486278]
- 10.
- Alarcon R A, Meienhofer J, Atherton E. Isophosphamide as a new acrolein-producing antineoplastic isomer of cyclophosphamide. Cancer Res. 1972;32:2519–2523. [PubMed: 5082597]
- 11.
- Lee S M, Crowther D, Scarffe J H. et al. Cyclophosphamide decreases O6-alkylguanine-DNA alkyltransferase activity in peripheral lymphocytes of patients undergoing bone marrow transplantation. Br J Cancer. 1992;66:331–336. [PMC free article: PMC1977821] [PubMed: 1387001]
- 12.
- Friedman H S, Pegg A E, Johnson S P. et al. Modulation of cyclophosphamide activity by O-6-alkylguanine-DNA alkyltransferase. Cancer Chemother Pharmacol. 1999;43:80–85. [PubMed: 9923545]
- 13.
- Russo JE, Hilton J, Colvin OM. The role of aldehyde dehydrogenase isoenzymes in cellular resistance to the alkylating agent cyclophosphamide. In Enzymology and Molecular Biology of Carbonyl Metabolism, vol 2. New York, Liss, 1989, p 65. [PubMed: 2726824]
- 14.
- Antman K H, Elias A, Ryan L. Ifosfamide and mesna: response and toxicity at standard- and high-dose schedules. Semin Oncol. 1990;17:68. [PubMed: 2110386]
- 15.
- Loehrer P J Sr, Lauer R, Roth B J, Williams S D. et al. Salvage therapy in recurrent germ cell cancer: ifosfamide and cisplatin plus either vinblastine or etoposide. Ann Intern Med. 1988;109:540. [PubMed: 2844110]
- 16.
- Pratt C B, Douglass E C, Etcubanas E. et al. Clinical studies of ifosfamide/mesna at St. Jude Children’s Research Hospital, 1983–1988. Semin Oncol. 1989;16(Suppl 3):51. [PubMed: 2495567]
- 17.
- Boddy A V, Cole M, Pearson A D J, Idle J R. The kinetics of the auto-induction ifosfamide metabolism during continuous infusion. Cancer Chemother Pharmacol. 1995;36:53. [PubMed: 7720176]
- 18.
- Colvin M. The comparative pharmacology of cyclophosphamide and ifosfamide. Semin Oncol. 1982;9:2. [PubMed: 6761865]
- 19.
- Boal J H, Williamson M, Boyd V L, Ludeman S M, Egan W P. NMR studies of the kinetics of bisalkylation by isophosphoramide mustard: comparisons with phosphoramide mustard. J Med Chem. 1989;32:1768. [PubMed: 2754703]
- 20.
- Goren M P, Wright R K, Pratt C B, Pell F E. Dechlorethylation of ifosfamide and neurotoxicity. Lancet. 1986;2:1219. [PubMed: 2877353]
- 21.
- Costa G, Engle R L Jr, Schilling A. et al. Melphalan and prednisone: an effective combination for the treatment of multiple myeloma. Am J Med. 1973;54:589.
- 22.
- Barlogie B, Jagannath S, Dixon D O. et al. High-dose melphalan and granulocyte-macrophage colony-stimulating factor for refractory multiple myeloma. Blood. 1990;76:677. [PubMed: 2200536]
- 23.
- Frick J C, Tretter P, Tretter W, Hyman G A. Disseminated carcinoma of the ovary treated by L-phenylalanine mustard. Cancer. 1968;21:508. [PubMed: 5637955]
- 24.
- Young R C, Walton L A, Ellenberg S S. et al. Adjuvant therapy in stage I and stage II epithelial ovarian cancer. N Engl J Med. 1990;322:1021. [PubMed: 2181310]
- 25.
- Fisher B, Sherman B, Rockette H. et al. L-phenylalanine mustard (L-PAM) in the management of premenopausal patients with primary breast cancer. Cancer. 1979;44:847. [PubMed: 383274]
- 26.
- Rivkin S E, Green S, Metch B. et al. Adjuvant CMFVP versus melphalan for operable breast cancer with positive axillary nodes: 10-year results of a Southwest Oncology Group Study. J Clin Oncol. 1989;7:1229. [PubMed: 2671283]
- 27.
- Begleiter A, Lam H -Y P, Grover J, Froese E, Goldenberg G J. Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res. 1979;39:353. [PubMed: 570091]
- 28.
- Vistica D T, Rabon A, Rabinowitz M. Effects of l-alpha-amino-gamma-guanidinobutyric acid on melphalan therapy of the L1210 murine leukemia. Cancer Lett. 1979;6:345. [PubMed: 455272]
- 29.
- Vistica D T, Toal J N, Rabinowitz M. Amino acid conferred protection against melphalan: characterization of melphalan transport and correlation of uptake with cytotoxicity in cultured L1210 murine leukemia cells. Biochem Pharmacol. 1978;27:2865. [PubMed: 736979]
- 30.
- Greig N H, Momma S, Sweeney D J, Smith Q R, Rapoport S I. Facilitated transport of melphalan at the rat blood-brain barrier by the large neutral amino acid carrier system. Cancer Res. 1987;47:1571. [PubMed: 3815357]
- 31.
- Groothuis D R, Lippitz B E, Fekete I. et al. The effect of an amino acid-lowering diet on the rate of melphalan entry into brain and xenotransplanted glioma. Cancer Res. 1992;52:5590. [PubMed: 1394182]
- 32.
- The French Cooperative Group on Chronic Lymphocytic Leukemia.A randomized clinical trial or chlorambucil versus COP in stage B chronic lymphocytic leukemia. Blood. 1990;75:1422. [PubMed: 2180493]
- 33.
- Rundles R W, Striggle J, Bell W. et al. Comparison of chlorambucil and Myleran in chronic lymphocytic and granulocytic leukemia. Am J Med. 1959;27:424.
- 34.
- Harding M, Kennedy R, Mill L. et al. A pilot study of carboplatin (JM8, CBDCA) and chlorambucil in combination for advanced ovarian cancer. Br J Cancer. 1988;58:640. [PMC free article: PMC2246806] [PubMed: 3064798]
- 35.
- Wiltshaw E. Chlorambucil in the treatment of primary adenocarcinoma of the ovary. J Obstet Gynecol Br Commonw. 1964;72:586. [PubMed: 14341113]
- 36.
- Galton D A G, Israels L S, Nabarro J D N, Till M. Clinical trials of p-(di-2-chlorethylamino)-phenylbutyric acid (CB 1348) in malignant lymphoma. BMJ. 1955;2:172. [PMC free article: PMC1981252] [PubMed: 13269823]
- 37.
- Portlock C S, Fischer D S, Cadman E. et al. High-dose pulse chlorambucil in advanced, low-grade non-Hodgkin lymphoma. Cancer Treat Rep. 1987;71:1029. [PubMed: 3677110]
- 38.
- Greenspan E M. Thio-TEPA and methotrexate chemotherapy of advanced ovarian carcinoma. J Mt Sinai Hosp N Y. 1968;35:52. [PubMed: 4966488]
- 39.
- Perloff M, Hart R D, Holland J F. Vinblastine, adriamycin, thio-TEPA, and Holotestin (VATH). Cancer. 1978;42:2534. [PubMed: 103610]
- 40.
- Gutin P H, Levi J A, Wiernik P H, Walker M D. Treatment of malignant meningeal disease with intrathecal thioTEPA: a phase II study. Cancer Treat Rep. 1977;61:885. [PubMed: 408003]
- 41.
- Ng S F, Waxman D J. N,N’,N”-triethylenethiophosphoramide (thio-TEPA) oxygenation by constitutive hepatic P450 enzymes and modulation of drug metabolism and clearance in vivo by P450-inducing agents. Cancer Res. 1991;51:2340. [PubMed: 1707751]
- 42.
- Miller B, Teneholz T, Egorin M J. et al. Cellular pharmacology of N,N’,N”-triethylene thiophosphoramide. Cancer Lett. 1988;41:157. [PubMed: 3135933]
- 43.
- Hagen B, Neverdal G, Walstad R A, Nilsen O G. Long-term pharmacokinetics of thio-TEPA, TEPA and total alkylating activity following IV bolus administration of thioTEPA in ovarian cancer patients. Cancer Chemother Pharmacol. 1990;25:257. [PubMed: 1688514]
- 44.
- Heideman R L, Cole D E, Balis F. et al. Phase I and pharmacokinetic evaluation of thiotepa in the cerebrospinal fluid and plasma of pediatric patients: Evidence for dose-dependent plasma clearance of thiotepa. Cancer Res. 1989;49:736. [PubMed: 2491958]
- 45.
- Hagen B. Pharmacokinetics of thio-TEPA and TEPA in the conventional dose-range and its correlation to myelosuppressive effects. Cancer Chemother Pharmacol. 1991;27:373. [PubMed: 1705489]
- 46.
- Egorin M J, Snyder S W. Characterization of nonexchangeable radioactivity in L1210 cells incubated with (14C) thiotepa: labeling of phosphatidylethanolamine. Cancer Res. 1990;50:4044. [PubMed: 2112983]
- 47.
- Teicher B A, Waxman D J, Holden S A. et al. Evidence for enzymatic activation and oxygen involvement in cytotoxicity and antitumor activity of N,N’,N” triethylenethiophosphoramide. Cancer Res. 1989;49:4996. [PubMed: 2504483]
- 48.
- Lyss A P, Luedke S L, Einhorn L, Luedke D W, Raney M. Vindesine and mitomycin C in metastatic breast cancer. A Southeastern Cancer Study Group Trial. Oncology. 1989;46:357. [PubMed: 2511534]
- 49.
- Menichetti E T, Silva R R, Tummarello D, Miseria S. et al. Etoposide and mitomycin-C in pretreated metastatic breast cancer. Tumori. 1989;75:473. [PubMed: 2513674]
- 50.
- Wils J, Bleiberg H. Current status of chemotherapy for gastric cancer. Eur J Cancer Clin Oncol. 1989;25:3. [PubMed: 2646133]
- 51.
- Dorr R T, Bowden G T, Alberts D S, Liddil J D. Interactions of mitomycin C with mammalian DNA detected by alkaline elution. Cancer Res. 1985;45:3510. [PubMed: 3926301]
- 52.
- Kennedy K A, McGuirl J D, Leondaridis L, Alabaster O. pH dependence of mitomycin C-induced cross-linking activity in tumor cells. Cancer Res. 1985;45:3541. [PubMed: 3926302]
- 53.
- Borowy-Borowski H, Lipman R, Chowdary D, Tomasz M. Duplex oligodeoxyribonucleotides cross-linked by mitomycin C at a single site: synthesis, properties, and cross-link reversibility. Biochemistry. 1990;29:2992. [PubMed: 2110820]
- 54.
- Borowy-Borowski H, Lipman R, Tomasz M. Recognition between mitomycin C and specific DNA sequences for cross-link formation. Biochemistry. 1990;29:2999. [PubMed: 2110821]
- 55.
- Iyer V N, Szybalski W. Mitomycin and porfiromycin: chemical mechanisms of activation and cross-linking of DNA. Science. 1964;145:55. [PubMed: 14162693]
- 56.
- Khan A S, Driscoll J S. Potential central nervous system antitumor agents. J Med Chem. 1976;19:313. [PubMed: 1249812]
- 57.
- Curt G A, Kelley J A, Kufta C V. et al. Phase II and pharmacokinetic study of aziridinylbenzoquinone (2,5-diazirindinyl-3, 6-bis(carboethoxyamino)-1, 4-benzoquinone, diaziquone, NSC 182986) in high-grade gliomas. Cancer Res. 1983;43:6102. [PubMed: 6640549]
- 58.
- Falletta J M, Cushing B, Lauer S. et al. Phase I evaluation of diaziquone in childhood cancer. A Pediatric Oncology Group study. Invest New Drugs. 1990;8:167. [PubMed: 2384303]
- 59.
- Gutierrez P L. Mechanism(s) of bioreductive activation. The example of diaziquone (AZQ). Free Radic Biol Med. 1989;6:405. [PubMed: 2651223]
- 60.
- Ross D, Siegel D, Gibson N W. et al. Activation and deactivation of quinones catalyzed by DT-diaphorase. Evidence for bioreductive activation of diaziquone (AZQ) in human tumor cells and detoxification of benzene metabolites in bone marrow stroma. Free Radic Res Commun. 1990;8:373. [PubMed: 2113030]
- 61.
- Chiuten D F, Rosensweig M, Von Hoff D D, Muggia F M. Clinical trials with hexitol derivatives in the U.S. Cancer. 1981;47:442. [PubMed: 6784907]
- 62.
- Haas C D, Stephens R C, Hollister M, Hoogstraten B. Phase I evaluation of dianhydrogalactitol (NSC-132313). Cancer Treat Rep. 1976;60:611. [PubMed: 991150]
- 63.
- Tormey D C, Falkson G, Simon R M. A randomized comparison of two sequentially administered regimens to a single regimen in metastatic breast cancer. Cancer Clin Trials. 1979;2:247.
- 64.
- Sellei C, Ecklardt S, Horvath I P, Kralovanszky J, Institoris L. Clinical and pharmacologic experience with dibromodulcitol (NSC-104800), a new antitumor agent. Cancer Chemother Rep. 1969;53:377. [PubMed: 4915082]
- 65.
- Haddow A, Timmis G M. Myleran in chronic myeloid leukemia-chemical constitution and biological action. Lancet. 1953;1:207. [PubMed: 13097986]
- 66.
- Pacheco D Y, Stratton N K, Gibson N W. Comparison of the mechanism of action of busulfan with hepsulfam, a new antileukemic agent, in the L1210 cell line. Cancer Res. 1989;49:5108. [PubMed: 2766282]
- 67.
- Elson L A. Hematological effects of the alkylating agents. Ann N Y Acad Sci. 1958;68:826. [PubMed: 13627735]
- 68.
- Fried W, Kede A, Barone J. Effects of cyclophosphamide and busulfan on spleen-colony-forming units and on hematopoietic stroma. Cancer Res. 1977;37:1205. [PubMed: 321118]
- 69.
- Galton D. Myleran in chronic myeloid leukaemia. Lancet. 1953;1:208. [PubMed: 13234407]
- 70.
- Galton D A G, Till M, Wiltshaw E. Busulfan (1, 4-dimethyl-sulfonoxy-butane, Myleran): summary of clinical results. Ann N Y Acad Sci. 1958;68:967. [PubMed: 13627743]
- 71.
- Santos G W, Tutschka P J, Brookmeyer R. et al. Marrow transplantation for acute non-lymphocytic leukemia after treatment with busulfan and cyclophosphamide. N Engl J Med. 1983;309:1347. [PubMed: 6355849]
- 72.
- Linker C A, Ries C A, Damon L E. et al. Autologous bone marrow transplantation for acute myeloid leukemia using 4-hydroperoxy-cyclophosphamide purged bone marrow and the busulfan/etoposide preparative regimen: a follow-up report. Bone Marrow Transplant. 1998;22:865–872. [PubMed: 9827814]
- 73.
- Hyde K A, Acton E, Skinner W A. et al. Potential anticancer agents-LX11. The relationship of chemical structure to antileukemia activity with analogues of 1-methyl-3-nitro-1-nitrosoguanidine (NSC-9369). II. J Med Pharm Chem. 1962;5:1. [PubMed: 14046609]
- 74.
- Schabel F M Jr, Johnston T P, McCaleb G S. et al. Experimental evaluation of potential anticancer agents: VIII. Effects of certain nitrosoureas on cerebral L1210 leukemia. Cancer Res. 1963;23:226. [PubMed: 13976561]
- 75.
- DeVita V T, Carbone P P, Owens A H Jr. et al. Clinical trials with 1,3-bis (2-chloroethyl)-1-nitrosourea, NSC-409962. Cancer Res. 1965;25:1876. [PubMed: 5858571]
- 76.
- Colvin M, Brundrett R B, Cowens W, Jardine E, Ludlum D B. A chemical basis for the antitumor activity of chloroethylnitrosoureas. Biochem Pharmacol. 1976;25:695. [PubMed: 945062]
- 77.
- Kohn K W. Interstrand cross-linking of DNA by 1,3-bis(2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res. 1977;37:1450. [PubMed: 851960]
- 78.
- Ludlum D B, Kramer B S, Wang J. et al. Reaction of 1,3-bis(2-chloroethyl)-1 nitrosourea with synthetic polynucleotides. Biochemistry. 1975;14:5480. [PubMed: 1201275]
- 79.
- Walker M D, Alexander E Jr, Hunt W E. et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333. [PubMed: 355604]
- 80.
- Presant C A, Klahr C. Adriamycin, 1,3-bis-(2-chloroethyl)-1 -nitrosourea (BCNU, NSC No. 409962), cyclophosphamide plus prednisone (ABC-P) in melphalan resistant multiple myeloma. Cancer. 1978;42:1222–1227. [PubMed: 698913]
- 81.
- Schabel F M Jr. Nitrosoureas: a review of experimental anti-tumor activity. Cancer Treat Rep. 1976;60:665. [PubMed: 782694]
- 82.
- Krouwer D, McDermott M, Prados M. Postoperative radiotherapy and radiotherapy combined with CCNU chemotherapy for treatment of brain gliomas. J Neurooncol. 1990;8:189. [PubMed: 2358851]
- 83.
- Lefkowitz I B, Packer R J, Sielgel K R. et al. Results of treatment of children with recurrent medulloblastoma/primitive neuroectodermal tumors with lomustine, cisplatin, and vincristine. Cancer. 1990;65:412. [PubMed: 2153428]
- 84.
- Jackson D V Jr, Craig J B, Spurr C L. et al. Vincristine infusion with CHOP-CCNU in diffuse large-cell lymphoma. Cancer Invest. 1990;8:7. [PubMed: 2190676]
- 85.
- Lennard A L, Carey P J, Jackson G H, Proctor S J. An effective oral combination in advanced relapsed Hodgkins disease: prednisolone, etoposide, chlorambucil and CCNU. Cancer Chemother Pharmacol. 1990;26:301. [PubMed: 2369796]
- 86.
- Clark J L, Barcewicz P, Nava H R, Goodwin P S, Douglass H O Jr. Adjuvant 5-FU and MeCCNU improves survival following curative gastrectomy for adenocarcinoma. Am Surg. 1990;56:423. [PubMed: 2368986]
- 87.
- Gerard A, Metzger U, Buyse M. Adjuvant therapy in colorectal cancer. Anticancer Res. 1989;9:1033. [PubMed: 2683989]
- 88.
- Levin V A, Chamberlain M, Silver P, Rodriguez L, Prados M. Phase I/II study of intraventricular and intrathecal ACNU for leptomeningeal neoplasia. Cancer Chemother Pharmacol. 1989;23:301. [PubMed: 2706735]
- 89.
- Roosen N, Kiwit J C, Lins E, Schirmer M, Bock W J. Adjuvant intra-arterial chemotherapy with nimustine in the management of World Health Organization Grade IV gliomas of the brain. Cancer. 1984;64:1989. [PubMed: 2553234]
- 90.
- Japan Radiation-ACNU Study Group.A randomized prospective study of radiation versus radiation versus plus ACNU in inoperable non-small cell carcinoma of the lung. Cancer. 1989;63:249. [PubMed: 2535953]
- 91.
- Jacquillat C, Khayat D, Banzet P. et al. Chemotherapy by fotemustine in cerebral metastases of disseminated malignant melanoma. Cancer Chemother Pharmacol. 1990;25:263. [PubMed: 2403853]
- 92.
- Tapiero H, Yin M B, Catalin J. et al. Cytotoxicity and DNA damaging effects of a new nitrosourea, fotemustine, diethyl-1-(3-[2-chloroethyl]-3-nitrosoureido) ethylphosphonate-S10036. Anticancer Res. 1989;9:1617. [PubMed: 2627116]
- 93.
- Hartley-Asp B, Christensson P I, Gunnarsson K. et al. Anti-tumour, toxicological and pharmacokinetic properties of a novel taurine-based nitrosourea (TCNU). Invest New Drugs. 1988;6:19. [PubMed: 3410663]
- 94.
- Poisson M, Chiras J, Fauchon F, Debussche C, Delattre J Y. Treatment of malignant recurrent glioma by intra-arterial infra-ophthalmic infusion of HECNU 1-(2-chloroethyl)-1-nitroso-3-(2-hydroxyethyl) urea. A phase II study. J Neurooncol. 1990;8:255. [PubMed: 2193122]
- 95.
- Auerbuch SD. Nonclassic alkylating agents. In Cancer Chemotherapy: Principles and Practice. Edited by BA Chabner, JM Collins. Philadelphia: Lippincott, 1990, pp 314–328.
- 96.
- Erikson J M, Tweedie D J, Ducore J M, Prough R A. Cytotoxicity and DNA damage caused by the azoxy metabolites of procarbazine in L1210 tumor cells. Cancer Res. 1989;49:127. [PubMed: 2908840]
- 97.
- Swaffar D S, Horstman M G, Jaw J Y. et al. Methoxyprocarbazine, the active metabolite responsible for the anticancer activity of procarbazine against L1210 leukemia. Cancer Res. 1989;49:2442. [PubMed: 2706632]
- 98.
- Skibba J L, Beal D D, Ramirez G, Bryan T. N-Demethylation of the antineoplastic agent 4(5)-(3,3-dimethyl-1-triazeno)imidazole-5(4)-carboxamide by rats and man. Cancer Res. 1970;30:147. [PubMed: 5441073]
- 99.
- Vaughan K, Tang Y, Lianos G. et al. Studies of the mode of action of antitumor triazenes and trizines. 6. 1-Aryl-3(hydroxymethyl-3-methyltriazenes: synthesis, chemistry, and antitumor properties. J Med Chem. 1984;27:357. [PubMed: 6699881]
- 100.
- Bonnadonna G, Valgussa P, Santoro A. et al. Hodgkin’s disease: the Milan Cancer Institute experience with MOPP and ABVD. Recent Results Cancer Res. 1989;117:169. [PubMed: 2481328]
- 101.
- DeVita V T, Serpick A A, Carbone P P. Combination chemotherapy in the treatment of advanced Hodgkin’Æs disease. Ann Intern Med. 1970;73:881. [PubMed: 5525541]
- 102.
- Levin V A, Silver P, Hannigan J. et al. Superiority of post-radiotherapy adjuvant chemotherapy with CCNU, procarbazine, and vincristine (PCV) over BCNU for anaplastic gliomas. Int J Radiat Oncol Biol Phys. 1990;18:321. [PubMed: 2154418]
- 103.
- Flaherty L E, Redman B G, Chabot G G. et al. A phase I-II study of dacarbazine in combination with outpatient interleukin-2 in metastatic malignant melanoma. Cancer. 1990;65:2471. [PubMed: 2337862]
- 104.
- Kirkwood J M, Ernstoff M S, Giuliano A. et al. Interferon alpha-2a and dacarbazine in melanoma. J Natl Cancer Inst. 1990;82:1062. [PubMed: 2189999]
- 105.
- Auerbach, SD. Nonclassic alkylating agents. In Cancer Chemotherapy: Principles and Practice. Edited by BA Chabner, JM Collins. Philadelphia: Lippincott, 1990, pp 314–328.
- 106.
- Denny B J, Wheelhouse R T, Stevens M F, Tsang L L, Slack J A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry. 1994;33:9045. [PubMed: 8049205]
- 107.
- Newlands E S, Blackledge G R, Slack J A. et al. Phase I trial of temozolomide (CCRG 81045: m&B 39831: NSC 362856). Br J Cancer. 1992;65:287. [PMC free article: PMC1977719] [PubMed: 1739631]
- 108.
- Bower M, Newlands E S, Bleehen N M. et al. Multicentre Crc phase II trial of temozolomide in recurrent or progressive high-grade glioma. Cancer Chemother Pharmacol. 1997;40:484–488. [PubMed: 9332462]
- 109.
- Brock C S, Newlands E S, Wedge S R. et al. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998;58:4363–4367. [PubMed: 9766665]
- 110.
- Miller K J, McGovern R M, Ames M M. Effect of a hepatic activation system on the antiproliferative activity of hexamethylmelamine against human tumor cell lines. Cancer Chemother Pharmacol. 1985;15:49. [PubMed: 3924426]
- 111.
- Rutty C J, Abel G. In vitro cytotoxicity of the methylmelamines. Chem Biol Interact. 1980;29:235. [PubMed: 6766359]
- 112.
- Ross D, Langdon S P, Gescher A, Stevens M F. Studies of the mode of action of antitumour triazenes and triazines-V. The correlation of the in vitro cytotoxicity and in vivo antitumor activity of hexamethylmelamine analogues with their metabolism. Biochem Pharmacol. 1984;33:1131. [PubMed: 6424683]
- 113.
- Rutty C J, Judson I R, Abel G. et al. Preclinical toxicology, pharmacokinetics and formulation of N2,N4,N6-trihydroxymethyl-N2,N4,N6-trimethylmelamine (trimelamol), a water-soluble cytoxic s-triazine which does not require metabolic activation. Cancer Chemother Pharmacol. 1986;17:251. [PubMed: 3091280]
- 114.
- Meer L, Schold S C, Kleihues P. Inhibition of the hepatic 06-alkylguanine-DNA alkyltransferase in vivo by pretreatment with antineoplastic agents. Biochem Pharmacol. 1989;38:929. [PubMed: 2930593]
- 115.
- Greco F A, Johnson D H, Hainsworth J D. A comparison of hexamethylmelamine (altretamine), cyclophosphamide, doxorubicin, and cisplatin (H-CAP) vs. cyclophosphamide, cisplatin (CAP) in advanced ovarian cancer. Cancer Res. 1991;18(Suppl A):47. [PubMed: 1904309]
- 116.
- Hill D L, Kirk M C, Struck R F. Microsomal metabolism of nitrosoureas. Cancer Res. 1975;35:296. [PubMed: 234031]
- 117.
- Levin V A, Stearns J, Byrd A, Finn A, Weinkam R J. The effect of phenobarbital on the antitumor activity of 1,3-bis(2-chlorethyl)-1-nitrosourea (BCNU), 1-(2-chlorethyl)-3-cyclohexyl-1-nitrosourea (CCNU) and 1-(2-chlorethyl)-3-(2, 6-dioxo)-3-piperidyl-1-nitrosourea (PCNU), and on the plasma pharmacokinetics and biotransformation of BCNU. J Pharmacol Exp Ther. 1979;208:1. [PubMed: 759602]
- 118.
- Alberts D S, Chang S Y, Chen H -S G, Larcom B J, Jones S E. Pharmacokinetics and metabolism of chlorambucil in man: a preliminary report. Cancer Treat Rev. 1979;6(Suppl):9. [PubMed: 498174]
- 119.
- McLean A, Woods R C, Catovsky D, Farmer P. Pharmacokinetics and metabolism of chlorambucil in patients with maligant disease. Cancer Treat Rev. 1979;6(Suppl):33. [PubMed: 498170]
- 120.
- Sharma M, He Q Y, Tomasz M. Effects of glutathione on alkylation and cross-linking of DNA by mitomycin C. Isolation of a ternary glutathione-mitomycin-DNA adduct. Chem Res Toxicol. 1994;7:401. [PubMed: 8075372]
- 121.
- Loveless A, Ross W C J. Chromosome alteration and tumour inhibition by nitrogen mustards: the hypothesis of cross-linking alkylation. Nature. 1950;166:111. [PubMed: 14796714]
- 122.
- Brookes P, Lawley P D. The reaction of mono-and difunctional alkylating agents with nucleic acids. Biochem J. 1961;80:486. [PMC free article: PMC1243259] [PubMed: 16748923]
- 123.
- Brookes P, Lawley P D. The action of alkylating agents on deoxyribonucleic acid in relation to biologic effects of the alkylating agents. Exp Cell Res. 1963;9(Suppl):512. [PubMed: 14046250]
- 124.
- Ewig R A G, Kohn K W. DNA damage and repair in mouse leukemia L1210 cells treated with nitrogen mustard. 1,3-bis(2-chloroethyl)-1-nitrosourea, and other nitrosoureas. Cancer Res. 1977;37:2114. [PubMed: 558823]
- 125.
- Millard J T, Raucher S, Hopkins P B. Mechlorethamine cross links deoxyguanosine residues at 59 GNC sequences in duplex DNA fragments. J Am Chem Soc. 1990;112:2459.
- 126.
- JD, Grueneberg D A, Loechler E L. Synthesis of a duplex oligonucleotide containing a nitrogen mustard interstrand DNA-DND cross-link. Cancer Res. 1989;49:6529. [PubMed: 2819709]
- 127.
- Dong Q, Barsky D, Colvin M E. et al. A structural basis for a phosphoramide mustard-induced DNA interstrand cross-link at 5’-d(GAC). Proc Natl Acad Sci U S A. 1995;92:12170–12174. [PMC free article: PMC40318] [PubMed: 8618865]
- 128.
- Price C C, Gaucher G M, Koneru P. et al. Relative reactivities for monofunctional nitrogen mustard alkylation of nucleic acid components. Biochim Biophys Acta. 1968;166:327. [PubMed: 5680596]
- 129.
- Erickson L C, Laurent G, Sharkey N A, Kohn K W. DNA cross-linking and monoadduct repair in nitrosourea-treated human tumour cells. Nature. 1980;288:727. [PubMed: 7005689]
- 130.
- Parker S, Kirk M C, Ludlum D B. Synthesis and characterization of 0-6-(2-chloroethyl)guanine: a putative intermediate in the cytotoxic reaction of chloroethylnitrosoureas with DNA. Biochem Biophys Res Commun. 1987;148:1124. [PubMed: 3689390]
- 131.
- Bodell W J, Tokuda K, Ludlum D B. Differences in DNA alkylation products formed in sensitive and resistant human glioma cells treated with N-(2-chloroethyl)-N-nitrosurea. Cancer Res. 1988;48:4489. [PubMed: 3396000]
- 132.
- MacFarland J G, Kirk M C, Ludlum D B. Mechanism of action of the nitrosoureas. IV. Synthesis of the 2-haloethylnitrosourea-induced DNA cross-link 1-(3-cytosinyl),2-(1-guanyl)ethane. Biochem Pharmacol. 1990;39:33. [PubMed: 2297360]
- 133.
- Fischhaber P l, Gall A S, Duncan J A, Hopkins P B. Direct demonstration in synthetic oligonucleotides that N,N ‘-bis(2-chloroethyl)-nitrosourea cross-links N-1 of deoxyguanosine to N-3 of deoxycytidine on opposite strands of duplex DNA. Cancer Res. 1999;59:4363–4368. [PubMed: 10485484]
- 134.
- Verly W G. Monofunctional alkylating agents and apurinic sites in DNA. Biochem Pharmacol. 1974;23:3. [PubMed: 4590108]
- 135.
- Verly W G, Paquette Y. An endonuclease for depurinated DNA in Escherichia coli B. Can J Biochem. 1972;50:217–224. [PubMed: 4552312]
- 136.
- Kat A, Thilly W G, Fang W H. et al. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A. 1993;90:6424. [PMC free article: PMC46944] [PubMed: 8341649]
- 137.
- Duckett D R, Drummond J T, Murchie A I H. et al. Human MutS-apha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci U S A. 1996;93:6443–6447. [PMC free article: PMC39042] [PubMed: 8692834]
- 138.
- Goldenberg G J, Vanstone C L, Israels L G, Lise D, Bihler I. Evidence for a transport carrier of nitrogen mustard in nitrogen mustard-sensitive and -resistant L51784 lymphoblasts. Cancer Res. 1970;30:2285. [PubMed: 5475476]
- 139.
- Wolpert M K, Ruddon R W. A study on the mechanisms of resistance to nitrogen mustard (HN2) in Ehrlich ascites tumor cells: comparison of uptake of HN2-14C. Cancer Res. 1969;29:873. [PubMed: 5775714]
- 140.
- Dantzig A H, Fairgrieve M, Slayman C W, Adelberg E A. Isolation and characterization of a CHO amino acid transport mutant resistant to melphalan (L-phenylalanine mustard). Somat Cell Mol Genet. 1984;10:113. [PubMed: 6584987]
- 141.
- Redwood W R, Colvin M. Transport of melphalan by sensitive and resistant L1210 cells. Cancer Res. 1980;40:1144. [PubMed: 7357545]
- 142.
- Hilton J. Role of aldehyde dehydrogenase in cyclophosphamide-resistant L1210 leukemia. Cancer Res. 1984;44:5156. [PubMed: 6488175]
- 143.
- Koelling T M, Yeager A M, Hilton J, Haynie D T, Wiley J M. Development and characterization of a cyclophosphamide-resistant subline of acute myeloid leukemia in the Lewis and Brown Norway hybrid rat. Blood. 1990;76:1209. [PubMed: 2400809]
- 144.
- Colvin M, Russo J E, Hilton J, Dulik D M, Fenselau C. Enzymatic mechanisms of resistance to alkylating agents in tumor cells and normal tissues. Adv Enzyme Regul. 1988;27:211–221. [PubMed: 3074628]
- 145.
- Parsons P G, Lean J, Kable E P W. et al. Relationship between resistance to cross-linking agents and glutathione metabolism, aldehyde dehydrogenase isozymes and adenovirus replication in human tumor all lines. Biochem Pharmacol. 1990;40:2641. [PubMed: 2260988]
- 146.
- Rekha G K, Sreerama L, Sladek N E. Intrinsic cellular resistance to oxazaphosphorines exhibited by a human colon carcinoma cell line expressing relatively large amounts of a class-3 aldehyde dehydrogenase. Biochem Pharmacol. 1994;48:1943. [PubMed: 7986206]
- 147.
- Sreerama L, Sladek N E. Identification of a methylcholanthrene-induced aldehyde dehydrogenase in a human breast adenocarcinoma cell line exhibiting oxazaphosphorine-specific acquired resistance. Cancer Res. 1994;54:2176. [PubMed: 8174125]
- 148.
- Calcutt G, Conners T A. Tumor sulfhydryl levels and sensitivity to the nitrogen mustard merophan. Biochem Pharmacol. 1963;12:839. [PubMed: 14071541]
- 149.
- Friedman H S, Colvin O M, Kaufmann S H, Ludeman S H, Bullock N, Bigner D D, Griffith O W. Cyclophosphamide resistance in medulloblastoma. Cancer Res. 1992;52:5373. [PubMed: 1356617]
- 150.
- Suzukaka D, Petro B J, Vistica D T. Reduction in glutathione content of L-PAM resistant L1210 cell confers drug sensitivity. Biochem Pharmacol. 1982;31:121–124. [PubMed: 7059344]
- 151.
- Butler A L, Clapper M L, Tew K D. Glutathione S-transferase in nitrogen mustard-resistant and -sensitive cell lines. Mol Pharmacol. 1987;31:575. [PubMed: 3600602]
- 152.
- Nakagawa K, Saijo N, Tsuchida S. et al. Glutathione-S-transferase p as a determinant of drug resistance in transfectant cell lines. J Biol Chem. 1990;265:4296. [PubMed: 2407735]
- 153.
- Puchalski R B, Fahl W E. Expression of recombinant glutathione-S-transferase pi, Ya or Yb1 confers resistance to alkylating agents. Proc Natl Acad Sci U S A. 1990;87:2443. [PMC free article: PMC53705] [PubMed: 2320566]
- 154.
- Tew K D, Bomber A M, Hoffman S J. Ethacrynic acid and piriprost as enhancers of cytotoxicity in drug resistant and sensitive cell lines. Cancer Res. 1988;48:3622. [PubMed: 3288331]
- 155.
- Batist G, Behrens B C, Makuch R. et al. Serial determinations of glutathione levels and glutathione-related enzyme activities in human tumor cells in vitro. Biochem Pharmacol. 1986;35:2257. [PubMed: 3827990]
- 156.
- Ciaccio P J, Tew K D, Lacreta F P. The spontaneous and glutathione S-transferase mediated reaction of chlorambucil with glutathione. Cancer Commun. 1990;2:279. [PubMed: 2390420]
- 157.
- Friedman H S, Colvin O M, Aisaka K. et al. Glutathione protects cardiac and skeletal muscle from cyclophosphamide-induced toxicity. Cancer Res. 1990;50:2455. [PubMed: 2317829]
- 158.
- Mulcahy R T, Untawale S, Gipp J J. Transcriptional unregulation of gamma-glutamyl-cysteine synthetase gene expression in melphalan-resistant human prostate carcinoma cells. Mol Pharmacol. 1994;46:909. [PubMed: 7969079]
- 159.
- Dulik D M, Colvin O M, Fenselau C. Characterization of glutathione conjugates of chlorambucil by fast atom bombardment and thermospray liquid chromatography/mass spectrometry. Biomed Environ Mass Spectrom. 1990;19:248. [PubMed: 2340362]
- 160.
- Dulik D M, Fenselau C, Hilton J. Characterization of melphalan-glutathione adducts whose formation is catalyzed by glutathione S-transferase. Biochem Pharmacol. 1986;35:3405. [PubMed: 3768029]
- 161.
- Yuan Z M, Fenselau C, Dulik D M. et al. Laser desorption electron impact: application to a study of the mechanism of conjugation of glutathione and cyclophosphamide. Anal Chem. 1990;62:868. [PubMed: 2350000]
- 162.
- Bolton M G, Colvin O M, Hilton J. Specificity of isozymes of murine hepatic glutathione S-transferase for the conjugation of glutathione with L-phenylalanine mustard. Cancer Res. 1991;51:2410. [PubMed: 2015603]
- 163.
- Ciaccio P J, Tew K D, Lacreta F P. The spontaneous and glutathione S-transferase mediated reaction of chlorambucil with glutathione. Cancer Commun. 1990;2:279. [PubMed: 2390420]
- 164.
- Dirven H A, van Ommen B, van Bladeren P J. Involvement of human glutathione S-transferase isoenzymes in the conjugation of cyclophosphamide metabolites with glutathione. Cancer Res. 1994;54:6215. [PubMed: 7954469]
- 165.
- Pallante S L, Lisek C A, Dulik D M, Fenselau C. Glutathione conjugates Immobilized enzyme synthesis and characterization by fast atom bombardment mass spectrometry. Drug Metab Dispos. 1986;14:313. [PubMed: 2872031]
- 166.
- Smith M T, Evans C G, Doane-Setzer P. et al. Denitrosation of 1,3-bis(2-chlorethyl)-1-nitrosourea by class mu glutathione transferases and its role in cellular resistance in rat brain tumor cells. Cancer Res. 1989;49:2621. [PubMed: 2713846]
- 167.
- Friedman H S, Colvin O M, Griffith O W. et al. Increased melphalan activity in intracranial human medulloblastoma and glioma xenografts following buthionine sulfoximine-mediated glutathione depletion. J Natl Cancer Inst. 1989;81:524. [PubMed: 2921776]
- 168.
- Kramer R A, Greene K, Ahmad S, Vistica D T. Chemosensitization of L-phenylalaline mustard by the thiol-modulating agent buthionine sulfoximine. Cancer Res. 1987;47:1593. [PubMed: 3815359]
- 169.
- Ozols R F, Louie K G, Plowman J. et al. Enchanced melphalan cytotoxicity in human ovarian cancer in vitro and in tumor-bearing nude mice by buthionine sulfoximine depletion of glutathione. Biochem Pharmacol. 1987;36:147. [PubMed: 3801051]
- 170.
- Ishikawa M, Sasaki K, Takayanagi Y. Injurious effect of buthionine sulfoximine, an inhibitor of glutathione biosynthesis, on the lethality and urotoxicity of cyclophosphamide in mice. Jpn J Pharmacol. 1989;51:146. [PubMed: 2810938]
- 171.
- Smith A C, Liao J T, Page J G, Wientjes M G, Grieshaber C K. Pharmacokinetics of buthionine sulfoximine (NSC 326231) and its effect on melphalan-induced toxicity in mice. Cancer Res. 1989;49:5385. [PubMed: 2766304]
- 172.
- Bailey H H, Mulcahy R T, Tutsch K D. et al. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: An attempt at modulation of glutathione. J Clin Oncol. 1994;12:194. [PubMed: 8270977]
- 173.
- Bailey H H. L-S,R-buthionine sulfoximine: historical development and clinical issues. Chem Biol Interact. 1998;111-112:239–254. [PubMed: 9679558]
- 174.
- Bailey H H, Ripple G, Tutsch K D. et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 1997;89:1789–1796. [PubMed: 9392620]
- 175.
- Rhodes T, Twentyman P R. A study of ethacrynic acid as a potential modifier of melphalan and cisplatin sensitivity in human lung cancer parental and drug-resistant cell lines. Br J Cancer. 1992;65:684. [PMC free article: PMC1977394] [PubMed: 1316774]
- 176.
- Gupta V, Jani J P, Jacobs S. et al. Activity of melphalan in combination with glutathione transferase inhibitor sulfasalazine. Cancer Chemother Pharmacol. 1995;36:13. [PubMed: 7720170]
- 177.
- Andrews P A, Murphy M P, Howell S B. Metallothionein mediated cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol. 1987;19:149. [PubMed: 3568272]
- 178.
- Endresen L, Schjerven L, Rugstad H E. Tumours from a cell strain with a high content of metallothionein show enhanced resistance against cis-dichlorodiammineplatinum. Acta Pharmacol Toxicol (Copenh). 1984;55:183. [PubMed: 6542297]
- 179.
- Kelley S L, Basu A, Teicher B A. et al. Overexpression of metallothionein confers resistance to anticancer drugs. Science. 1988;241:1813. [PubMed: 3175622]
- 180.
- Yu X, Wu Z, Fenselau C. Covalent sequestration of melphalan by metallothionein alkylation of cysteines. Biochemistry. 1995;34:3377. [PubMed: 7880833]
- 181.
- Wei D, Fabris D, Fenselau C. Covalent sequestration of phosphoramide mustard by metallothionein—an in vitro study. Drug Metab Dispos. 1999;27:786–791. [PubMed: 10383921]
- 182.
- Pegg A E. Mammalian 06-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119. [PubMed: 2205376]
- 183.
- Ali-Osman F, Srivenugopal K, Berger M S, Stein D E. DNA interstrand cross-linking and strand break repair in human glioma cell lines of varying (1,3-bis(2-chlorethyl)-1-nitrosourea) resistance. Anticancer Res. 1990;10:677. [PubMed: 2164350]
- 184.
- Felker G M, Friedman H S, Dolan M E, Moschel R C, Schold C. Treatment of subcutaneous and intracranial brain tumor xenografts with 0-6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Chemother Pharmacol. 1993;32:471. [PubMed: 8258196]
- 185.
- Schold S C Jr, Brent T P, von Hofe E. et al. 06-alkylguanine-DNA alkyltransferase and sensitivity to procarbazine in human brain-tumor xenografts. J Neurosurg. 1989;70:573. [PubMed: 2926498]
- 186.
- Brent T P, Houghton P J, Houghton J A. 06-Alkylguanine-DNA alkyltransferase activity correlates with the therapeutic response of human rhabdomyosarcoma xenografts to 1-(2-chlorethyl)-3-(trans-4-methylcyclohexyl)-1-nitrosourea. Proc Natl Acad Sci U S A. 1985;82:2985. [PMC free article: PMC397691] [PubMed: 3857628]
- 187.
- Cussac C, Rapp M, Mounetou E. et al. Enhancement by O6-benzyl-N-acetylguanosine derivatives of chloroethylnitrosourea antitumor action in chloroethylnitrosourea-resistant human malignant melanocytes. J Pharmacol Exp Ther. 1994;271:1353. [PubMed: 7996446]
- 188.
- Magull-Seltenreich A, Zeller W J. Inhibition of 06-alkylguanine-DNA alkyltransferase in animal and human ovarian tumor cell lines by 06-benzylguanine and sensitization to BCNU. Cancer Chemother Pharmacol. 1995;35:262. [PubMed: 7805187]
- 189.
- Futscher B W, Micetich K C, Barnes D M, Fisher R I, Erickson L C. Inhibition of specific DNA repair system and nitrosourea cytotoxicity in resistant human cancer cells. Cancer Commun. 1989;1:65. [PubMed: 2534817]
- 190.
- Wedge S R, Porteus J K, May B l. et al. Potentiation of temozolomide and BCNU cytotoxicity by O(6)-benzylguanine: a comparative study in vitro. Br J Cancer. 1996;73:482–490. [PMC free article: PMC2074446] [PubMed: 8595163]
- 191.
- Mitchell R B, Moschel R C, Dolan M E. Effect of 0-6-benzylguanine on the sensitivity of human tumor xenografts to 1,3-bis(2-chloroethyl)-1-nitrosourea and on DNA interstrand cross-link formation. Cancer Res. 1992;52:1171. [PubMed: 1737376]
- 192.
- Pegg A E, Boosalis M, Samson M. et al. Mechanism of inactivation of human 06-alkylaguanine-DNA alkyltransferase by 06-benzylguanine. Biochemistry. 1993;32:11998. [PubMed: 8218276]
- 193.
- Spiro T p, Gerson S L, Liu L L. et al. O-6-benzylguanine: a clinical trial establishing the biochemical modulatory dose in tumor tissue for alkyltransferase-directrd DNA repair. Cancer Res. 1999;59:2402–2410. [PubMed: 10344750]
- 194.
- Dolan M E, Roy S K, Fasanmade A A. et al. O-6-Benzylguanine in humans—metabolic, pharmacokinetic, and pharmacodynamic findings. J Clin Oncol. 1998;16:1803–1810. [PubMed: 9586894]
- 195.
- Friedman H S, Kokkinakis D M, Pluda J. et al. Phase I trial of O-6-benzylguanine for patients undergoing surgery for malignant glioma. J Clin Oncol. 1998;16:3570–3575. [PubMed: 9817277]
- 196.
- Davis B M, Reese J S, Kocet O. et al. Selection for G156A O6-methylguanine DNA methyltransferase gene-transduced hematopoietic progenitors and protection from lethality in mice treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1997;57:5093–5099. [PubMed: 9371508]
- 197.
- Xu-Welliver M, Kanugula S, Pegg A E. et al. Isolation of human O-6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O-6-benzylguanine. Cancer Res. 1936;58:1936–1945. [PubMed: 9581836]
- 198.
- Kohn K W, Steigbigel N H, Spears C L. Cross-linking and repair of DNA in sensitive and resistant strains of E. coli treated with nitrogen mustard. Proc Natl Acad Sci U S A. 1965;53:1154. [PMC free article: PMC301387] [PubMed: 5330356]
- 199.
- Dong Q, Bullock N, Ali-Osman F. et al. Repair analysis of 4-hydroperoxycyclo-phosphamide induced DNA interstrand crosslinking in the c-myc gene In 4-hydroperoxycyclophamide-sensitive and -resistant medulloblastoma cell lines. Cancer Chemother Pharmacol. 1996;37:242–246. [PubMed: 8529284]
- 200.
- Stevnsner T, Ding R, Smulson M, Bohr V A. Inhibition of gene-specific repair of alkylation damage in cells depleted of poly(ADP-ribose) polymerase. Nucleic Acids Res. 1994;22:4620. [PMC free article: PMC308509] [PubMed: 7984410]
- 201.
- Connor P M, Ferris D K, White G A. et al. Relationships between cdc2 kinase DNA cross-linking, and cell cycle perturbations induced by nitrogen mustards. Cell Growth Diff. 1992;3:43. [PubMed: 1534688]
- 202.
- Das S K, Lau C C, Pardee A B. Comparative analysis of caffeine and 3-aminobenzamide as DNA repair inhibitors in Syrian baby hamster kidney cells. Mutat Res. 1984;131:71. [PubMed: 6700619]
- 203.
- Selby C P, Sancar A. Molecular mechanisms of DNA repair inhibition by caffeine. Proc Natl Acad Sci U S A. 1990;87:3522. [PMC free article: PMC53933] [PubMed: 2185474]
- 204.
- van Zeeland A A, Bussmann C J, Degrassi F. et al. Effects of aphidicolin on repair replication and induced chromosomal aberrations in mammalian cells. Mutat Res. 1982;92:379. [PubMed: 6806654]
- 205.
- Teicher B A, Herman T S, Holden S A. et al. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science. 1990;247:1457. [PubMed: 2108497]
- 206.
- Green S k, Frankel A, Kerbel R S. Adhesion-dependent multicellular drug resistance. Anti Cancer Drug Design. 1999;14:153–168. [PubMed: 10405642]
- 207.
- Kobayashi H, Man S, Graham C H. et al. Acquired multicellular-mediated resistance to alkylating agents in cancer. Proc Natl Acad Sci U S A. 1993;90:3294. [PMC free article: PMC46286] [PubMed: 8475071]
- 208.
- Graham C H, Kobayashi H, Stankiewicz K S. et al. Rapid acquisition of multicellular drug resistance after a single dose of antitumor alkylating agents. J Natl Cancer Inst. 1994;86:953. [PubMed: 8007019]
- 209.
- Kerbel R S, Kobayashi H, Graham C H. Intrinsic or acquired drug resistance and metastasis: are they linked phenotypes? J Cell Biochem. 1994;56:37. [PubMed: 7806590]
- 210.
- Jahde E, Glusenkamp K H, Rajewsky M F. Protection of cultured malignant cells from mitoxantrone cytotoxicity by low extracellular pH: a possible mechanism for chemoresistance in vivo. Eur J Cancer. 1990;26:101. [PubMed: 2138903]
- 211.
- Egorin M J, Forrest A, Belani C P. et al. A limited sampling strategy for cyclophosphamide pharmacokinetics. Cancer Res. 1989;49:3129. [PubMed: 2720671]
- 212.
- Jardine I, Fenselau C, Appler M. et al. Quantitation by gas chromatography-chemical ionization mass spectrometry of cyclophosphamide, phosphamide mustard, and nornitrogen mustard in the plasma and urine of patients receiving cyclophosphamide therapy. Cancer Res. 1978;38:408. [PubMed: 620410]
- 213.
- Struck R F, Alberts D S, Horne K. et al. Plasma pharmacokinetics of cyclophosphamide and its cytotoxic metabolites after intravenous versus oral administration in a randomized, crossover trial. Cancer Res. 1987;47:2723. [PubMed: 3552204]
- 214.
- Jao J Y, Jusko W J, Cohen J L. Phenobarbital effects on cyclophosphamide pharmacokinetics in man. Cancer Res. 1972;32:2761. [PubMed: 4678592]
- 215.
- D’Incalci M, Bolis G, Facchinetti T. et al. Decreased half-life of cyclophosphamide in patients under continual treatment. Eur J Cancer. 1979;15:7. [PubMed: 421718]
- 216.
- Erlichman C, Soldins S J, Hardy R W, Thiessen J J, Sturgeon J F, Fine S, Baskerville T. Disposition of cyclophosphamide on two consecutive cycles of treatment in patients with ovarian carcinoma. Arzneimittelforschung. 1988;38:839. [PubMed: 3178926]
- 217.
- Sladek N. Therapeutic efficacy of cyclophosphamide as a function of its metabolism. Cancer Res. 1972;32:535. [PubMed: 5061308]
- 218.
- Chen T L, Passos-Coelho J L, Noe D A. et al. Nonlinear pharmacokinetics of cyclophosphamide in patients with metastatic breast cancer receiving high-dose chemotherapy followed by autologous bone marrow transplantation. Cancer Res. 1995;55:810. [PubMed: 7850794]
- 219.
- Anderson L W, Ludeman S M, Colvin O M, Grochow L B, Strong J M. Quantitation of 4-hydroxycyclophosphamide/aldophosphamide in whole blood. J Chromatogr B Biomed Appl. 1995;667:247–257. [PubMed: 7663697]
- 220.
- Chen T L, Kennedy M J, Anderson L W. et al. Nonlinear pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in patients with metastatic breast cancer receiving high-dose chemotherapy followed by autologous bone marrow transplantation. Drug Metab Dispos. 1997;25:544–551. [PubMed: 9152592]
- 221.
- Slattery J T, Kalhorn T F, McDonald G B. et al. Conditioning regimen-dependent disposition of cyclophosphamide and hydroxycyclophosphamide in human marrow transplantation patients. J Clin Oncol. 1996;14:1484–1494. [PubMed: 8622062]
- 222.
- Bakke J E, Feil V J, Fjelstul C E, Thacker E J. Metabolism of cyclophosphamide by sheep. J Agric Food Chem. 1972;20:384. [PubMed: 5016621]
- 223.
- Struck R F, Kirk M C, Mellett L B, El-Dareer S, Hill D L. Urinary metabolites of the antitumor agent cyclophosphamide. Mol Pharmacol. 1971;7:519. [PubMed: 5139563]
- 224.
- Humphrey R L, Kvols L K. The influence of renal insufficiency on cyclophosphamide-induced hematopoietic depression and recovery. Proc Am Assoc Cancer Res. 1974;15:84.
- 225.
- Brade W P, Herdrich K, Varini M. Ifosfamide pharmacology, safety and therapeutic potential. Cancer Treat Rev. 1985;12:1. [PubMed: 3896483]
- 226.
- Nelson R L, Allen L M, Creaven P J. Pharmacokinetics of divided-dose ifosfamide. Clin Pharmacol Ther. 1976;19:365. [PubMed: 1261170]
- 227.
- Norpoth K. Studies on the metabolism of isophosphamide (NSC-109724) in man. Cancer Treat Rep. 1976;60:437. [PubMed: 1277219]
- 228.
- Wagner T, Heydrich D, Jork T, Voelcker G, Hohorst H J. Comparative study on human pharmacokinetics of activated ifosfamide and cyclophosphamide by a modified fluorometric test. J Cancer Res Clin Oncol. 1981;100:95. [PubMed: 7240346]
- 229.
- Struck, RF, Alberts, DS, Horne K. et al. Plasma pharmacokinetics of cyclophosphamide and its cytotoxic metabolites after intravenous versus oral administration in a randomized, crossover, trial. Cancer Res. 1987;47:2723. [PubMed: 3552204]
- 230.
- Brain E G, Yu L J, Gustafsson K. et al. Modulation of P450-dependent ifosfamide pharmacokinetics: a better understanding of drug activation in vivo. Br J Cancer. 1998;77:1768–1776. [PMC free article: PMC2150313] [PubMed: 9667645]
- 231.
- Ducharme M P, Bernstein M l, Granvil C P. et al. Phenytoin-induced alteration in the N-dechloroethylation of ifosfamide stereoisomers. Cancer Chemother Pharmacol. 1997;40:531–533. [PubMed: 9332469]
- 232.
- Granvil C P, Madan A, Sharkawi M. et al. Role of CYP2B6 and CYP3A4 in the in vitro N-dechloroethylation of (R)- and (S)-ifosfamide in human liver microsomes. Drug Metab Dispos. 1999;27:533–541. [PubMed: 10101149]
- 233.
- Alberts D S, Chang S Y, Chen H -S G. et al. Kinetics of intravenous melphalan. Clin Pharmacol Ther. 1979;26:73. [PubMed: 445964]
- 234.
- Alberts D S, Chang S Y, Chen H -S G, Larcom B J, Evans T L. Comparative pharmacokinetics of chlorambucil and melphalan in man. Recent Results Cancer Res. 1980;74:124. [PubMed: 7444135]
- 235.
- Kergueris M F, Milpied N, Moreau P, Harousseau J L, Larousse C. Pharmacokinetics of high-dose melphalan in adults: influence of renal function. Anticancer Res. 1994;14:2379. [PubMed: 7825976]
- 236.
- Pallante S L, Fenselau C, Mennel R G. et al. Quantitation by gas chromatography-chemical ionization-mass spectrometry of phenylalanine mustard in plasma of patients. Cancer Res. 1980;40:2268. [PubMed: 7388793]
- 237.
- Choi K E, Ratain M J, Williams S F. et al. Plasma pharmacokinetics of high-dose oral melphalan in patients treated with trialkylator chemotherapy and autologous bone marrow. Cancer Res. 1989;49:1318. [PubMed: 2645050]
- 238.
- Tattersall M H N, Weinberg A. Pharmacokinetics of melphalan following oral or intravenous administration in patients with malignant disease. Eur J Cancer. 1978;14:507. [PubMed: 648565]
- 239.
- Reece P A, Kotasek D, Morris R G, Dale B M, Sage R E. The effect of food on oral melphalan absorption. Cancer Chemother Pharmacol. 1986;16:194. [PubMed: 3948305]
- 240.
- Cornwell G G III, Pajak T F, McIntyre O R, Kochna S, Dosik H. Influence of renal failure on myelosuppressive effects of melphalan: cancer and leukemia group B experience. Cancer Treat Rep. 1982;66:475. [PubMed: 7060036]
- 241.
- Alberts D S, Chen H -S G, Benz D, Mason N L. Effect of renal dysfunction in dogs on the disposition and marrow toxicity of melphalan. Br J Cancer. 1981;43:330. [PMC free article: PMC2010602] [PubMed: 7225283]
- 242.
- Reece P A, Hill H S, Green R M. et al. Renal clearance and protein binding of melphalan in patients with cancer. Cancer Chemother Pharmacol. 1988;22:348. [PubMed: 3168148]
- 243.
- Hartvig P, Simonsson B, Oberg G, Wallin I, Ehrsson H. Inter- and intraindividual differences in oral chlorambucil pharmacokinetics. Eur J Clin Pharmacol. 1988;35:551. [PubMed: 3069479]
- 244.
- Cohen B E, Egorin M J, Kohlhepp E A, Aisner J, Gutierrez P L. Human plasma pharmacokinetics and urinary excretion of thiotepa and its metabolites. Cancer Treat Rep. 1986;70:859. [PubMed: 2424593]
- 245.
- Henner W D, Shea T C, Furlong E A. et al. Pharmacokinetics of continuous-infusion high-dose thiotepa. Cancer Treat Rep. 1987;71:1043. [PubMed: 3119200]
- 246.
- Wadler S, Egorin M J, Zuhowski E G. et al. Phase I clinical and pharmacokinetic study of thiotepa administered intraperitoneally in patients with advanced malignancies. J Clin Oncol. 1989;7:132. [PubMed: 2491884]
- 247.
- Strong J M, Collins J M, Lester C, Poplack D G. Pharmacokinetics of intraventricular and intravenous N,N’,N”-triethylenethiophosphoramide (thiotepa) in rhesus monkeys and humans. Cancer Res. 1986;46(12 Pt 1):6101. [PubMed: 3096555]
- 248.
- Levin V A, Hoffman W, Weinkam R J. Pharmacokinetics of BCNU in man: a preliminary study of 20 patients. Cancer Treat Rep. 1978;62:1305. [PubMed: 688274]
- 249.
- Henner W D, Peters W P, Eder J P. et al. Pharmacokinetics and immediate effects of high-dose carmustine in man. Cancer Treat Rep. 1986;70:877. [PubMed: 3719578]
- 250.
- Lee F Y, Workman P, Roberts J T, Bleehen N M. Clinical pharmacokinetics of oral CCNU (lomustine). Cancer Chemother Pharmacol. 1985;14:125. [PubMed: 3971475]
- 251.
- Chow D S, Bhagwatwar H P, Phadungpojna S. et al. Stability-indicating high-performance liquid chromatographic assay of busulfan in aqueous and plasma samples. J Chromatogr B Biomed Sci Appl. 1997;704:277–288. [PubMed: 9518161]
- 252.
- Grochow L B, Jones R J, Brundrett B R. et al. Pharmacokinetics of busulfan: correlation with veno-occulusive disease in patients undergoing bone marrow transplantation. Cancer Chemother Pharmacol. 1989;25:55. [PubMed: 2591002]
- 253.
- Hassan O G, Ehrsson H. et al. Pharmacokinetic and metabolic studies of high-dose busulfan in adults. Eur J Clin Pharmacol. 1989;36:525. [PubMed: 2753072]
- 254.
- Vassal G, Gouyette A, Hartmann O. et al. Pharmacokinetics of high-dose busulfan in children. Cancer Chemother Pharmacol. 1989;24:386. [PubMed: 2791192]
- 255.
- Grochow L B, Krivit W, Whitley C B, Blazar B. Busulfan disposition in children. Blood. 1990;75:1723. [PubMed: 2328321]
- 256.
- Grochow L B. Busulfan disposition: the role of therapeutic monitoring in bone mar-row transplantation induction regimens. Semin Oncol. 1993;20(Suppl 4):18. [PubMed: 8342071]
- 257.
- Vassal G. Pharmacologically-guided dose adjustment of busulfan in high-dose chemotherapy regimensùrationale and pitfalls. Anticancer Res. 1994;14:2363. [PubMed: 7825973]
- 258.
- Nissen-Meyer R, Host H. A comparison between the hematological side effects of cyclophosphamide and nitrogen mustard. Cancer Chemother. 1960;9:51. [PubMed: 13729268]
- 259.
- Mullins G M, Colvin M. Intensive cyclophosphamide therapy in solid tumors. Cancer Chemother Rep. 1975;59:411. [PubMed: 1097100]
- 260.
- Reyes E S, Talley R W, O’Bryan R M. et al. Clinical evaluation of 1,3-bis- (2-chloroethyl)-1-nitrosourea (BCNU; NSC-409962) with fluoxymesterone (NSC-12165) in the treatment of solid tumors. Cancer Chemother Rep. 1973;57:225. [PubMed: 4582564]
- 261.
- Talmadge J E, Tribble H, Pennington R. et al. Protective, restorative, and therapeutic properties of recombinant colony-stimulating factors. Blood. 1989;73:2093. [PubMed: 2471557]
- 262.
- Brandt S J, Peters W P, Atwater S K. et al. Effect of recombinant human granulocyte-macrophage colony-stimulating factor on hemotopoietic reconstitution after high-dose chemotherapy and autologous bone marrow transplantation. N Engl J Med. 1988;318:869. [PubMed: 3281007]
- 263.
- Gianni A M, Bregni M, Siena S. et al. Recombinant human granulocyte-macrophage colony-stimulating factor reduces hematologic toxicity and widens clinical applicability of high-dose cyclophosphamide treatment in breast cancer and non-Hodgkin’s lymphoma. J Clin Oncol. 1990;8:768. [PubMed: 2185337]
- 264.
- Taylor K M, Jagannath S, Spitzer G. et al. Recombinant human granulocyte colony-stimulating factor hastens granulocyte recovery after high-dose chemotherapy and autologous bone marrow transplantation in Hodgkin’s disease. J Clin Oncol. 1989;7:1791. [PubMed: 2479719]
- 265.
- Kobayashi S, Teramura M, Oshimi K, Mizoguchi H. Interleukin-11 [review] Leuk Lymphoma. 1994;15:45. [PubMed: 7532057]
- 266.
- Lok S, Foster D C. The structure, biology and potential therapeutic applications of recombinant thrombopoietin. Stem Cells. 1994;12:586. [PubMed: 7881359]
- 267.
- Eder J P, Antman K, Peters W. et al. High-dose combination alkylating agent chemotherapy with autologous bone marrow support for metastatic breast cancer. J Clin Oncol. 1986;4:1592. [PubMed: 3534155]
- 268.
- Antman K, Eder J P, Elias A. et al. High-dose thiotepa alone and in combination regimens with bone marrow support. Semin Oncol. 1990;17(Suppl 3):33. [PubMed: 2106166]
- 269.
- McElwain T J, Hedley D W, Gordon M Y. et al. High dose melphalan and non-cryopreserved autologous bone marrow treatment of malignant melanoma and neuroblastoma. Exp Hematol. 1979;7 Suppl 5:360. [PubMed: 400698]
- 270.
- Thatcher D, Lind M, Morgenstern G. et al. High-dose, double alkylating agent chemotherapy with DTIC, melphalan, or ifosfamide and marrow rescue for metastatic malignant melanoma. Cancer. 1989;63:1296. [PubMed: 2646005]
- 271.
- Borison H L, Brand E D, Orland R K. Emetic action of nitrogen mustard (Mechlorethamine hydrochloride) in dogs and cats. Am J Physiol. 1968;192:410. [PubMed: 13508892]
- 272.
- Fetting J H, McCarthy L E, Borison H L, Colvin M. Vomiting induced by cyclophosphamide and phosphoramide mustard in cats. Cancer Treat Rep. 1982;66:1625. [PubMed: 7105052]
- 273.
- Carden P A, Mitchell S L, Waters K D, Tiedemann K, Ekert H. Prevention of cyclophosphamide/cytarabine-induced emesis with ondansetron in children with leukemia. J Clin Oncol. 1990;8:1531. [PubMed: 2144019]
- 274.
- Gez E, Sulkes A, Ochayon L. et al. Methylprednisolone versus metoclopramide as antiemetic treatment in patients receiving adjuvant cyclophosphamide, methotrexate, 5-fluorouracil (CMF) chemotherapy: a randomized crossover blind study. J Chemother. 1989;1:365. [PubMed: 2693621]
- 275.
- Grunberg S M. Advances in the management of nausea and vomiting induced by non-cisplatin containing chemotherapeutic regimens. Blood Rev. 1989;3:216. [PubMed: 2692744]
- 276.
- Jones R J, Lee K S, Beschorner W E. et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation. 1987;44:778. [PubMed: 3321587]
- 277.
- Peters W P, Eder J P, Henner W D. et al. High-dose combination alkylating agents with autologous bone marrow support: a phase I trial. J Clin Oncol. 1986;4:646. [PubMed: 3517240]
- 278.
- Phillips G L, Fay J W, Herzig G P. et al. Intensive 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), NSC A4366650 and cryopreserved autologous marrow transplantation for refractory cancer. A Phase I-II study. Cancer. 1983;51:1792. [PubMed: 6354414]
- 279.
- Nimer S D, Milewicz A L, Champlin R E, Busittil R W. Successful treatment of hepatic venoocclusive disease in a bone marrow transplant patient with orthotopic liver transplantation. Transplantation. 1990;49:819. [PubMed: 2326879]
- 280.
- Rhodes D F, Lee W M, Wingard J R. et al. Orthotopic liver transplantation for graft-versus-host disease following bone marrow transplantation. Gastroenterology. 1990;99:536. [PubMed: 2365200]
- 281.
- Spitz S. The histological effects of nitrogen mustards on human tumors and tissues. Cancer. 1948;1:383. [PubMed: 18887905]
- 282.
- Miller D G. Alkylating agents and human spermatogenesis. JAMA. 1971;217:1662. [PubMed: 4937384]
- 283.
- Sherins R J, DeVita V T. Effect of drug treatment for lymphoma on male reproductive capacity. Ann Intern Med. 1973;79:216. [PubMed: 4580112]
- 284.
- Blake D B, Heller R H, Hsu S H, Schacter B Z. Return of fertility in a patient with cyclophosphamide-induced azoospermia. Johns Hopkins Med J. 1976;139:20. [PubMed: 948148]
- 285.
- Hinkes E, Plotkin D. Reversible drug-induced sterility in a patient with acute leukemia. JAMA. 1973;223:1490. [PubMed: 4513133]
- 286.
- Miller J J, Williams G F, Leissring J C. Multiple late complications of therapy with cyclophosphamide, including ovarian destruction. Am J Med. 1971;50:530. [PubMed: 5313910]
- 287.
- Rose D P, Davis T E. Ovarian function in patients receiving adjuvant chemotherapy for breast cancer. Lancet. 1977;1:1174. [PubMed: 68275]
- 288.
- Kyoma H, Wada T, Nishizawa T, Iwanaga T, Aoki Y. Cyclophosphamide-induced ovarian failure and its therapeutic significance in patients with breast cancer. Cancer. 1977;39:1403. [PubMed: 851940]
- 289.
- Oliner H, Schwartz R, Rubio F Jr, Dameshek W. Interstitial pulmonary fibrosis following busulfan therapy. Am J Med. 1961;31:134. [PubMed: 13730733]
- 290.
- Patel A R, Shah P C, Rhee H L, Sassoon H, Rao K P. Cyclophosphamide therapy and interstitial pulmonary fibrosis. Cancer. 1976;38:1542. [PubMed: 1068740]
- 291.
- Radin A E, Haggard M E, Travis L B. Lung changes and chemotherapeutic agents in childhood. Am J Dis Child. 1970;120:337. [PubMed: 5537047]
- 292.
- Bailey C C, Marsden H B, Jones P H. Fatal pulmonary fibrosis following 1,3-bis (2-chloroethyl)-1-nitrosourea (BCNU) therapy. Cancer. 1978;42:74. [PubMed: 667809]
- 293.
- Holoye P Y, Jenkins D E, Greenberg S D. Pulmonary toxicity in long-term administration of BCNU. Cancer Treat Rep. 1976;60:1691. [PubMed: 1021241]
- 294.
- Codling B W, Chakera T M. Pulmonary fibrosis following therapy with melphalan for multiple myeloma. J Clin Pathol. 1972;25:668. [PMC free article: PMC477474] [PubMed: 5076801]
- 295.
- Cole R C, Myers T J, Klatsky A U. Pulmonary disease with chlorambucil therapy. Cancer. 1978;41:455. [PubMed: 630532]
- 296.
- Orwoll E S, Kiessling P J, Patterson J R. Interstitial pneumonia from mitomycin. Ann Intern Med. 1978;89:352. [PubMed: 686548]
- 297.
- Todd N W, Peters W P, Ost A H, Roggli V L, Piantadosi C A. Pulmonary drug toxicity in patients with primary breast cancer treated with high-dose combination chemotherapy and autologous bone marrow transplantation. Am Rev Resp Dis. 1993;147:1264. [PubMed: 8484641]
- 298.
- Forni A M, Koss L G, Geller W. Cytological study of the effect of cyclophosphamide on the epithelium of the urinary bladder in man. Cancer. 1964;17:1348. [PubMed: 14236768]
- 299.
- Philips F S, Sternberg S S, Cronin A P, Vidal P M. Cyclophosphamide and urinary bladder toxicity. Cancer Res. 1961;21:1577. [PubMed: 14486208]
- 300.
- Cox P J. Cyclophosphamide cystitis-identification of acrolein as the causative agent. Biochem Pharmacol. 1979;28:2045. [PubMed: 475846]
- 301.
- Van Dyk J J, Falkson H C, Van der Merwe A M, Falkson G. Unexpected toxicity in patients treated with iphosphamide. Cancer Res. 1972;32:921. [PubMed: 4552918]
- 302.
- Brock N. The development of mesna for the inhibition of urotoxic side effects of cyclophosphamide, ifosfamide, and other oxazaphosphorine cytostatics. Recent Results Cancer Res. 1980;74:270. [PubMed: 6777836]
- 303.
- Pratt C B, Horowitz M E, Meyer W H. et al. Phase II trial of ifosfamide in children with malignant solid tumors. Cancer Treat Rep. 1987;71:131. [PubMed: 3100034]
- 304.
- DeFronzo R A, Braine H G, Colvin M, Davis P J. Water intoxication in man after cyclophosphamide therapy. Ann Intern Med. 1973;78:861. [PubMed: 4713567]
- 305.
- Bode U, Seif S M, Levine A A. Studies on the antidiuretic effect of cyclophosphamide: vasopressin release and sodium excretion. Med Pediatr Oncol. 1980;8:295. [PubMed: 7464688]
- 306.
- Harlow P J, DeClerck Y A, Shore N A. et al. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44:896. [PubMed: 476599]
- 307.
- Green T P, Mirkin B L. Prevention of cyclophosphamide-induced antidiuresis by furosemide infusion. Clin Pharmacol Ther. 1981;29:634. [PubMed: 7214794]
- 308.
- Harmon W E, Cohen H J, Schneeberger E E, Grupe W E. Chronic renal failure in children treated with methyl CCNU. N Engl J Med. 1979;300:1200. [PubMed: 431647]
- 309.
- Schacht R G, Baldwin D S. Chronic interstitial nephritis and renal failure due to nitrosourea (NU) therapy. Kidney Int. 1978;14:661.
- 310.
- Kashimura M, Kondo M, Abe T, Shinohara M, Baba S. A case report of acute renal failure induced by melphalan in a patient with ovarian cancer. Gan No Rinsho. 1988;34:2015. [PubMed: 2849690]
- 311.
- Bierman H R, Kelly K H, Knudson A G Jr, Maekawa T, Timmis G M. The influence of 1, 4-dimethylsulfonoxy-1, 4-dimethylbutane (CB 2348, di-methyl Myleran) in neoplastic disease. Ann N Y Acad Sci. 1958;68:1211. [PubMed: 13627774]
- 312.
- Ganci L, Serrou B. Changes in hair pigmentation associated with cancer chemotherapy. Cancer Treat Rep. 1980;64:193. [PubMed: 7379056]
- 313.
- Feil V S, Lamoureaux C J H. Alopecia activity of cyclophosphamide metabolites and related compounds in sheep. Cancer Res. 1974;34:2596. [PubMed: 4411919]
- 314.
- Cornwell G G III, Pajak T F, McIntyre O R. Hypersensitivity reactions to IV melphalan during treatment of multiple myeloma: cancer and leukemia Group B experience. Cancer Treat Rep. 1979;63:399. [PubMed: 427822]
- 315.
- Lakin J D, Cahill R A. Generalized urticaria to cyclophosphamide: type I hypersensitivity to an immunosuppressive agent. J Allergy Clin Immunol. 1976;58:160. [PubMed: 985658]
- 316.
- Ross W E, Chabner B A. Allergic reaction to cyclophosphamide in a mechlorethamine-sensitive patient. Cancer Treat Rep. 1977;61:495. [PubMed: 872148]
- 317.
- Karchmer R K, Hansen B L. Possible anaphylactic reaction to intravenous cyclophosphamide. JAMA. 1977;237:475. [PubMed: 576273]
- 318.
- Kim H C, Kesarwala H H, Colvin M, Saidi P. Hypersensitivity reaction to a metabolite of cyclophosphamide. J Allergy Clin Immunol. 1985;76:591. [PubMed: 4056247]
- 319.
- Buckner C D, Rudolph R H, Fefer A. et al. High dose cyclophosphamide therapy for malignant disease. Cancer. 1972;29:357.
- 320.
- Slavin R E, Millan J C, Mullins G M. Pathology of high dose intermittent cyclophosphamide therapy. Hum Pathol. 1975;6:693. [PubMed: 1183993]
- 321.
- Steinherz L J, Steinherz P G. Cyclophosphamide cardiotoxicity. Cancer Bull. 1985;37:231.
- 322.
- Appelbaum F, Strauchen J A, Graw R G Jr. et al. Acute lethal carditis caused by high-dose combination chemotherapy: a unique clinical and pathological entity. Lancet. 1976;1:58. [PubMed: 54581]
- 323.
- Steinberg S S, Philips F S, Scholler J. Pharmacological and pathological effects of alkylating agents. Ann N Y Acad Sci. 1958;68:811. [PubMed: 13627734]
- 324.
- Bethlenfalvay N C, Bergin J J. Severe cerebral toxicity after intravenous nitrogen mustard therapy. Cancer. 1972;29:366. [PubMed: 5013538]
- 325.
- Eder J P, Elias A, Shea T C. et al. A phase I-II study of cyclophosphamide, thiotepa, and carboplatin with autologous bone marrow transplantation in solid tumor patients. J Clin Oncol. 1990;8:1239. [PubMed: 2162912]
- 326.
- Takvorian T, Parker L M, Hochberg F H. et al. Single high-dose of BCNU with autologous bone marrow (ABM). Proc AACR Am Soc Clin Oncol. 1980;21:341.
- 327.
- Yamada K, Bremer A M, West C R. et al. Intra-arterial BCNU therapy in the treatment of metastatic brain tumor from lung carcinoma. Cancer. 1979;44:2000. [PubMed: 509386]
- 328.
- Vassal G, Deroussent A, Hartmann O. et al. Dose-dependent neurotoxicity of high-dose-busulfan in children: a clinical and pharmacological study. Cancer Res. 1990;50:6203. [PubMed: 2400986]
- 329.
- Bodenstein D, Goldin A. A comparison of the effects of various nitrogen mustard compounds on embryonic cells. J Exp Zool. 1948;108:75. [PubMed: 18863655]
- 330.
- Murphy M L, Del Moro A, Lacon C. The comparative effects of five poly-functional alkylating agents on the rat fetus, with additional notes. Ann N Y Acad Sci. 1958;68:762. [PubMed: 13627731]
- 331.
- Gibson J E, Becker B A. Teratogenicity of structural truncates of cyclophosphamide in mice. Teratology. 1971;4:141.
- 332.
- Hales B F. Effects of phosphoramide mustard and acrolein, cytotoxic metabolites of cyclophosphamide, on mouse limb development in vitro. Teratology. 1989;40:11. [PubMed: 2763206]
- 333.
- Little S A, Mirkes P E. DNA cross-linking and single-strand breaks induced by teratogenic concentrations of 4-hydroperoxycyclophosphamide and phosphoramide mustard in postimplantation rat embryos. Cancer Res. 1987;47:5421. [PubMed: 3477317]
- 334.
- Mirkes P E. Cyclophosphamide teratogenesis: a review. Teratog Carcinog Mutagen. 1985;5:75. [PubMed: 2859667]
- 335.
- Garrett M J. Teratogenic effects of combination chemotherapy. Ann Intern Med. 1974;80:667. [PubMed: 4823823]
- 336.
- Steege J F, Caldwell D S. Renal agenesis after first trimester exposure to chlorambucil. South Med J. 1980;73:1414. [PubMed: 7434066]
- 337.
- Toledo T M, Harper R C, Moser R H. Fetal effects during cyclophosphamide and irradiation therapy. Ann Intern Med. 1971;74:87. [PubMed: 5539280]
- 338.
- Nicholson H O. Cytotoxic drugs in pregnancy. J Obstet Gynaecol Br Commonw. 1968;75:307. [PubMed: 4868587]
- 339.
- Lergier J E, Jiminez E, Maldonado N. et al. Normal pregnancy in multiple myeloma treated with cyclophosphamide. Cancer. 1974;34:1018. [PubMed: 4417641]
- 340.
- Ortega J. Multiple agent chemotherapy including bleomycin of non-Hodgkin’s lymphoma during pregnancy. Cancer. 1977;40:2829. [PubMed: 73411]
- 341.
- Hochberg M C, Shulman L E. Acute leukemia following cyclophosphamide therapy for Sjogren’s syndrome. Johns Hopkins Med J. 1978;142:211. [PubMed: 275517]
- 342.
- Kyle R A, Pierce R V, Bayrd E D. Multiple myeloma and acute myelomonocytic leukemia. N Engl J Med. 1970;283:1121. [PubMed: 5273282]
- 343.
- Rosner F, Grunwald H. Multiple myeloma terminating in acute leukemia. Am J Med. 1974;57:927. [PubMed: 4611209]
- 344.
- Rosner F, Grunwald H. Hodgkins disease and acute leukemia. Am J Med. 1975;58:339. [PubMed: 1090158]
- 345.
- Einhorn N. Acute leukemia after chemotherapy (melphalan). Cancer. 1978;41:444. [PubMed: 272946]
- 346.
- Reimer R R, Hoover R, Fraumeni J F Jr, Young R C. Acute leukemia after alkylating-agent therapy of ovarian cancer. N Engl J Med. 1977;297:177. [PubMed: 406560]
- 347.
- Tucker M A, Coleman C N, Cox R S, Varghese A, Rosenberg S A. Risk of second cancers after treatment for Hodgkins disease. N Engl J Med. 1988;318:76. [PubMed: 3336397]
- 348.
- Dorr F A, Coltman C A Jr. Second cancers following antineoplastic therapy. Curr Probl Cancer. 1985;9:1. [PubMed: 3888536]
- 349.
- Green M H, Harris E L, Gershenson D M. et al. Melphalan may be a more potent leukemogen than cyclophosphamide. Ann Intern Med. 1986;105:360. [PubMed: 3740675]
- 350.
- Einhorn N, Eklund G, Lambert B. Solid tumours and chromosome aberrations as late side effects of melphalan therapy in ovarian carcinoma. Acta Oncol. 1988;27:215. [PubMed: 3415849]
- 351.
- Penn I. Second malignant neoplasm associated with immunosuppressive medications. Cancer. 1976;37:1024. [PubMed: 766952]
- 352.
- Hektoen L, Corper H J. The effect of mustard gas (dichloroethyl-sulphide) on antibody formation. J Infect Dis. 1921;28:279.
- 353.
- Makinodan T, Santos G W, Quinn R P. Immunosuppressive drugs. Pharmacol Rev. 1970;22:189. [PubMed: 4914001]
- 354.
- Barratt T M, Soothill J F. Controlled trial of cyclophosphamide in steroid-sensitive relapsing nephrotic syndrome of childhood. Lancet. 1970;2:479. [PubMed: 4194935]
- 355.
- Laros R K Jr, Penner J A. “Refractory” thrombocytopenic purpura treated successfully with cyclophosphamide. JAMA. 1971;215:445. [PubMed: 5107390]
- 356.
- Townes A S, Sowa J M, Schuman L E. Controlled trial of cyclophosphamide in rheumatoid arthritis (RA): an 11-month double-blind crossover study. Arthritis Rheum. 1972;15:129.
- 357.
- Santos G W, Sensenbrenner L L, Anderson P N. et al. HLA-identical marrow transplants in aplastic anemia, acute leukemia, and lymphosarcoma employing cyclophosphamide. Transplant Proc. 1976;8:607. [PubMed: 11591]
- 358.
- Berd D, Mastrangelo M J. Active immunotherapy of human melanoma exploiting the immunopotentiating effects of cyclophosphamide. Cancer Invest. 1988;6:337. [PubMed: 3167614]
- 359.
- Dray S, Mokyr M B. Cyclophosphamide and melphalan as immunopotentiating agents in cancer therapy. Med Oncol Tumor Pharmacother. 1989;6:77. [PubMed: 2657252]
- 360.
- Ozer H, Cowens J W, Colvin M, Nussbaum-Blumenson A, Sheedy D. In vitro effects of 4-hydroperoxycyclophosphamide on human immunoregulatory T subset function. 1. Selective effects on lymphocyte function in T-B cell collaboration. J Exp Med. 1982;155:276. [PMC free article: PMC2186561] [PubMed: 6976414]
- 361.
- Berd D, Mastrangelo M J. Effect of low dose cyclophosphamide on the immune system of cancer patients: depletion of CD41, 2H41 suppressor-inducer T-cells. Cancer Res. 1988;48:1671. [PubMed: 2830969]
- 362.
- Mitchell M S, Kempf R A, Harel W. et al. Effectiveness and tolerability of low-dose cyclophosphamide and low-dose intravenous interleukin-2 in disseminated melanoma. J Clin Oncol. 1988;6:409. [PubMed: 2965219]
- 363.
- Mullins G M, Anderson P N, Santos G W. High dose cyclophosphamide therapy in solid tumors. Cancer. 1975;36:1950. [PubMed: 128408]
- 364.
- Rosenberg B, Van Camp L, Krigas T. Nature 1965;98.
- 365.
- Rosenberg B, Van Camp L, Trosko J E, Mansour V H. Plantinum compounds: a new class of potent antitumor agents. Nature. 1969;222:385. [PubMed: 5782119]
- 366.
- Higby D J, Wallace H J Jr, Albert D J, Holland J F. Diaminodichloroplatinum: a phase I study showing responses in testicular and other tumors. Cancer. 1974;33:1219. [PubMed: 4856724]
- 367.
- Higby D J, Wallace H J Jr, Holland J F. Cis-diamminedichloroplatinum (NSC-119875): a phase I study. Cancer Chemother Rep. 1973;57:459. [PubMed: 4586953]
- 368.
- Lippman A J, Helson C, Helson L, Krakoff I H. Clinical trials of cis-diamminedichloroplatinum (NSC-119875). Cancer Chemother Rep. 1973;57:191. [PubMed: 4126381]
- 369.
- Forastiere A A. Overview of platinum chemotherapy in head and neck cancer. Semin Oncol. 1994;21(5 Suppl 12):20. [PubMed: 7527591]
- 370.
- Ozols R F. Carboplatin and Taxol (paclitaxel) in advanced ovarian carcinoma. Ann Oncol. 1994;5 Suppl 6:S39. [PubMed: 7865433]
- 371.
- Pfeiffer P, Bennedbaek O, Bertelsen K. Intraperitoneal carboplatin in the treatment of minimal residual ovarian cancer. Gynecol Oncol. 1990;36:306. [PubMed: 2180794]
- 372.
- Skarlos D V, Samantas E, Kosmidis P. et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Cooperative Oncology Group Study. Ann Oncol. 1994;5:601. [PubMed: 7993835]
- 373.
- Viren M, Liippo K, Ojala A. et al. Carboplatin and etoposide in extensive small cell lung cancer. Acta Oncol. 1994;33:921. [PubMed: 7818926]
- 374.
- Grunberg S M, Sonka S, Stevenson L L, Muggia F M. Progressive paresthesias after cessation of therapy with very high-dose cisplatin. Cancer Chemother Pharmacol. 1989;25:62. [PubMed: 2556219]
- 375.
- Blatter E E, Vollano J E, Krishnan B S, Dabrowiak J C. Interaction of the antitumor agents cis,cis,trans-Pt(NH3)C12(OH)2 and cis,cis,trans-Pt IV((CH3)2CHNH2)2Cl2(OH)2 and their reduction products with Pmg DNA. D1. Biochemistry. 1984;23:4817. [PubMed: 6541947]
- 376.
- Pendyala L, Cowens J W, Chheda G B, Dutta S P, Creaven P J. Identification of cis-dichloro-bis-isopropylamine platinum(II) as a major metabolite of iproplatin in humans. Cancer Res. 1988;48:3533. [PubMed: 3370646]
- 377.
- Pendyala L, Walshm J R, Huq M M. et al. Uptake and metabolism of iproplatin in murine L1210 cells. Cancer Chemother Pharmacol. 1989;25:15. [PubMed: 2590997]
- 378.
- Hartley FR. The Chemistry of Platinum and Palladium, Chapter 11. New York: Wiley, 1973.
- 379.
- Martin RB. Hydrolytic equilibria and N7 versus N1 binding in purine nucleosides of cis-Diamminedichloroplatinum(II). In Platinum, Gold, and Other Metal Chemotherapeutic Agents. Edited by SJ Lippard. Washington, DC: American Chemical Society, 1983, p 231.
- 380.
- Roberts JJ, Pera MF. DNA as a target for anticancer coordination compounds. In Platinum, Gold, and the Metal Chemotherapeutic Agents. Edited by SJ Lippard. Washington, DC: American Chemical Society, 1983, p 3.
- 381.
- Zwelling L A, Anderson T, Kohn K W. DNA-protein and DNA interstrand cross-linking by cis- and trans-platinum(II) diamminedichloride in L1210 mouse leukemia cells and relation to cytotoxicity. Cancer Res. 1979;39:365. [PubMed: 570092]
- 382.
- Eastman A. Characterization of the adducts produced in DNA by cis-Diaminedichloroplatinum(II) and cis-Dichloro (ethylenediamine) platinum(II). Biochemistry. 1983;22:3927. [PubMed: 6225458]
- 383.
- Eastman A. Re-evaluation of interaction of cis-dichloro (ethylenediamine) platinum(II) with DNA. Biochemistry. 1986;25:3912. [PubMed: 3741840]
- 384.
- Fichtinger-Schepman A M J, van der Veer J L. et al. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985;24:707. [PubMed: 4039603]
- 385.
- Plooy A C, van Dijk M, Berends F, Lohman P H. Formation and repair of DNA interstrand cross-links in relation to cytotoxicity and unscheduled DNA synthesis induced in control and mutant human cells treated with cis-diamminedichloroplatinum(II). Cancer Res. 1985;45:4178. [PubMed: 3928152]
- 386.
- Knox R J, Friedlos F, Lydall D A, Roberts J J. Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato) platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res. 1986;46:1972. [PubMed: 3512077]
- 387.
- Poirier M C, Egorin M J, Fichtinger-Schepman A M. et al. DNA adducts of cisplatin and carboplatin in tissues of cancer patients. IARC Sci Publ. 1988;89:313. [PubMed: 3058599]
- 388.
- Sundquist W I, Lippard S J, Stollar B D. Monoclonal antibodies to DNA modified with cis- or trans-diamminedichloroplatinum(II). Proc Natl Acad Sci U S A. 1987;84:8225. [PMC free article: PMC299514] [PubMed: 2446320]
- 389.
- Pinto A L, Lippard S J. Sequence-dependent termination of in vitro DNA synthesis by cis- and trans-diamminedichloroplatinum(II). Proc Natl Acad Sci U S A. 1985;82:4616. [PMC free article: PMC390436] [PubMed: 3895221]
- 390.
- Villani G, Hubscer U, Butour J L. Sites of termination of in vitro DNA synthesis on cis-diamminedichloroplatinum treated single stranded DNA: a comparison between Escherichia coli DNA polymerase I and eucaryotic DNA polymerases alpha. Nucleic Acids Res. 1988;16:4407. [PMC free article: PMC336638] [PubMed: 3132699]
- 391.
- Heiger-Bernays W J, Essigmann J M, Lippard S J. Effect of the antitumor drug cis-diamminedichloroplatinum(II) and related platinum complexes on eukaryotic DNA replication. Biochemistry. 1990;29:8461. [PubMed: 2174701]
- 392.
- Sorenson C M, Eastman A. Influence of cis-diamminedichloroplatinum(II) on DNA synthesis and cell cycle progression in excision repair proficient and deficient Chinese hamster ovary cells. Cancer Res. 1988;48:6703. [PubMed: 3180081]
- 393.
- Drummond J T, Anthoney A, Brown R, Modrich P. Cisplatin and adriamycin resistance are associated with MutL-alpha and mismatch repair deficiency in an ovarian tumor cell line. J Biol Chem. 1996;271:19645–19648. [PubMed: 8702663]
- 394.
- Aebi S, Kurdi-Haidar B, Gordon R. et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed: 8674066]
- 395.
- Mello J A, Acharya S, Fishel R. et al. The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem Biol. 1996;3:579–589. [PubMed: 8807890]
- 396.
- Vaisman A, Varchenko M, Umar A. et al. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–3585. [PubMed: 9721864]
- 397.
- Fink D, Nebel S, Aebi S. et al. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed: 8895738]
- 398.
- Takahara P M, Frederick C A, Lippard S J. Crystal structure of the anticancer drug cisplatin bound to duplex DNA. JACS. 1996;118:12309–12321.
- 399.
- Gelasco A, Lippard S J. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry. 1998;37:9230–9239. [PubMed: 9649303]
- 400.
- Andrews P A, Velury S, Mann S C, Howell S B. Cis-Diamminendichloroplatinum(II) accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Res. 1988;48:68. [PubMed: 3335000]
- 401.
- Hromas R A, North J A, Burns C P. Decreased cisplatin uptake by resistant L1210 leukemia cells. Cancer Lett. 1987;36:197. [PubMed: 3621151]
- 402.
- Loh S Y, Mistry P, Kelland L R. et al. Reduced drug accumulation as a major mechanism of acquired resistance to cisplatin in a human ovarian carcinoma cell line: circumvention studies using novel platinum(II) and (IV) ammine/amine complexes. Br J Cancer. 1992;66:1109. [PMC free article: PMC1978040] [PubMed: 1457352]
- 403.
- Metcalfe S A, Cain K, Hill B T. Possible mechanism for differences in sensitivity to cis-platinum in human prostate tumor cell lines. Cancer Lett. 1986;31:163. [PubMed: 3697960]
- 404.
- Teicher B A, Holden S A, Kelley M J. et al. Characterization of a human squamous carcinoma cell line resistant to cis-diamminedichloroplatinum(II). Cancer Res. 1987;47:388. [PubMed: 3539321]
- 405.
- Fujii R, Mutoh M, Niwa K. et al. Active efflux system for cisplatin in cisplatin-resistant human cells. Jpn J Cancer Res. 1994;85:426. [PMC free article: PMC5919474] [PubMed: 8200854]
- 406.
- Mann S C, Andrews P A, Howell S B. Comparison of lipid content, surface membrane fluidity, and temperature dependence of cis-diamminedichloroplatinum(II) accumulation in sensitive and resistant human ovarian carcinoma cells. Anticancer Res. 1988;8:1211. [PubMed: 3218956]
- 407.
- Sharma R P, Edwards I R. Cis-Platinum: subcellular distribution and binding to cytosolic proteins. Biochem Pharmacol. 1983;32:2665. [PubMed: 6313005]
- 408.
- Andrews P A, Murphy M P, Howell S B. Differential sensitization of human ovarian carcinoma and mouse L1210 cells to cisplatin and melphalan by glutathione depletion. Mol Pharmacol. 1986;30:643. [PubMed: 3785141]
- 409.
- Behrens B C, Hamilton T C, Masuda H. et al. Characterization of a cis-diaminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414. [PubMed: 3539322]
- 410.
- Eastman A. Cross-linking of glutathione to DNA by cancer chemotherapeutic platinum coordination complexes. Chem Biol Interact. 1987;61:241. [PubMed: 3568194]
- 411.
- Fram R J, Woda B A, Wilson J M, Robichaud N. Characterization of acquired resistance to cis-diamminedichloroplatinum(II) in BE human colon carcinoma cells. Cancer Res. 1990;50:72. [PubMed: 2293559]
- 412.
- Hospers G A, Mulder N H, de Jong B. et al. Characterization of a human small cell lung carcinoma cell line with acquired resistance to cis-diamminedichloroplatinum(II) in vitro. Cancer Res. 1988;48:6803. [PubMed: 2846161]
- 413.
- Hromas R A, Andrews P A, Murphy M P, Burns C P. Glutathione depletion reverses cisplatin resistance in murine L1210 leukemia cells. Cancer Lett. 1987;34:9. [PubMed: 3802072]
- 414.
- Bier H, Bergler W, Mende S, Ganzer U. Glutathione content and gamma-glutamyltranspeptidase activity in squamous cell head and neck cancer xenografts. Arch Otorhinolaryngol. 1988;245:166. [PubMed: 2902836]
- 415.
- de Vries E G, Jeijer C, Timmer-Bosscha H. et al. Resistance mechanisms in three human small cell lung cancer cell lines established from one patient during clinical follow up. Cancer Res. 1989;49:4175. [PubMed: 2545337]
- 416.
- Andrews P A, Murphy M P, Howell S B. Characterization of cisplatin-resistance COLO 316 human ovarian carcinoma cells. Eur J Cancer Clin Oncol. 1989;25:619. [PubMed: 2714338]
- 417.
- Andrews P A, Schiefer M A, Murphy M P, Howell S B. Enhanced potentiation of cisplatin cytotoxicity in human ovarian carcinoma cells by prolonged glutathione depletion. Chem Biol Interact. 1988;65:51. [PubMed: 3345573]
- 418.
- Hamilton T C, Winker M A, Louie K G. et al. Augmentation of adriamycin, melphalan, and cisplatin cytotoxicity in drug-resistant and sensitive human ovarian carcinoma cell lines by buthionine sulfoximine mediated glutathione depletion. Biochem Pharmacol. 1985;34:2583. [PubMed: 4040369]
- 419.
- Dedon P C, Borch R F. Characterization of the reactions of platinum antitumor agents with biologic and nonbiologic sulfur-containing nucleophiles. Biochem Pharmacol. 1987;36:1955. [PubMed: 2954556]
- 420.
- Nakagawa K, Yokota J, Wada M. et al. Levels of glutathione S-transferase pi mRNA in human lung cancer cell lines correlate with the resistance to cisplatin and carboplatin. Jpn J Cancer Res. 1988;79:301. [PMC free article: PMC5917487] [PubMed: 2836347]
- 421.
- Saburi Y, Nakagawa M, Ono M. et al. Increased expression of glutathione S-transferase gene in cis-diamminedichloroplatinum(II)-resistant variants of a Chinese hamster ovary cell line. Cancer Res. 1989;49:7020. [PubMed: 2479474]
- 422.
- Wang Y Y, Teicher B A, Shea T C. et al. Cross-resistance and glutathione-S-transferase-pi levels among four human melonoma cell lines selected for alkylating agent resistance. Cancer Res. 1989;49:6185. [PubMed: 2804968]
- 423.
- Andrews P A, Murphy M P, Howell S B. Metallothionein mediated cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol. 1987;19:149. [PubMed: 3568272]
- 424.
- Endresen L, Schjerven L, Rugstad H E. Tumours from a cell strain with a high content of metallothionein show enhanced resistance against cis-dichlorodiammineplatinum. Acta Pharmacol Toxicol (Copenh). 1984;55:183. [PubMed: 6542297]
- 425.
- Kelley S L, Basu A, Teicher B A. et al. Overexpression of metallothionein confers resistance to anticancer drugs. Science. 1988;241:1813. [PubMed: 3175622]
- 426.
- Naganuma A, Satoh M, Imura N. Prevention of lethal and renal toxicity of cis-diamminedichloroplatinum(II) by induction of metallothionein synthesis without compromising its antitumor activity in mice. Cancer Res. 1987;47:983. [PubMed: 3802104]
- 427.
- Kraker A, Schmidt J, Krezoski S, Petering D H. Binding of cis-dichlorodiammine platinum(II) to metallothionein in Ehrlich cells. Biochem Biophys Res Commun. 1985;130:786. [PubMed: 4040756]
- 428.
- Zelazowski A J, Garvey J S, Hoeschele J D. In vivo and in vitro binding of platinum to metallothionein. Arch Biochem Biophys. 1984;229:246. [PubMed: 6538400]
- 429.
- Farnworth P G, Hillcoat B L, Roos I A. Metallothionein induction in mouse tissues by cis-dichlorodiammineplatinum(II) and its hydrolysis products. Chem Biol Interact. 1989;69:319. [PubMed: 2731304]
- 430.
- Van Den Berg H W, Roberts J J. Post-replication repair of DNA in Chinese hamster cells treated with cis platinum(II) diamine dichloride. Enhancement of toxicity and chromosome damage by caffeine. Mutat Res. 1975;33:279. [PubMed: 1240592]
- 431.
- Fravel H N, Roberts J J. Excision repair of cis-diamminedichloroplatinum(II)-induced damage to DNA of Chinese hamster cells. Cancer Res. 1979;39:1793. [PubMed: 427811]
- 432.
- Chu G, Berg P. DNA cross-linked by cisplatin: a new probe for the DNA repair defect in xeroderma pigmentosum. Mol Biol Med. 1987;4:277. [PubMed: 3695939]
- 433.
- Dijt F J, Fichtinger-Schepman A M, Berends F, Reedijk J. Formation and repair of cisplatin-induced adducts to DNA in cultured normal and repair-deficient human fibroblasts. Cancer Res. 1988;48:6058. [PubMed: 3167856]
- 434.
- Hansson J, Wood R D. Repair synthesis by human cell extracts in DNA damaged by cis- and trans-diamminedichloroplatinum(II). Nucleic Acids Res. 1989;17:8073. [PMC free article: PMC334948] [PubMed: 2554251]
- 435.
- Poll E H, Abrahams P J, Arwert F, Eriksson A W. Host-cell reactivation of cis-diamminedichloroplatinum(II)-treated SV40 DNA in normal human, Fanconi anaemia and xeroderma pigmentosum fibroblasts. Mutat Res. 1984;132:181. [PubMed: 6096706]
- 436.
- Poll E H, Arwert F, Joenje H, Eriksson A W. Cytogenetic toxicity of antitumor platinum compounds in Fanconi’s anemia. Hum Genet. 1982;61:228. [PubMed: 6890942]
- 437.
- Chao C C, Lee Y L, Lin-Chao S. Phenotypic reversion of cisplatin resistance in human cells accompanies reduced host cell reactivation of damaged plasmid. Biochem Biophys Res Commun. 1990;170:851. [PubMed: 2116798]
- 438.
- Eder J P, Teicher B A, Holden S A, Cathcart K N, Schnipper L E. Novobiocin enhances alkylating agent cytotoxicity and DNA interstrand cross-links in a murine model. J Clin Invest. 1987;79:1524. [PMC free article: PMC424429] [PubMed: 3033027]
- 439.
- Katz E J, Andrews P A, Howell S B. The effect of DNA polymerase inhibitors on the cytoxicity of cisplatin in human ovarian carcinoma cells. Cancer Commun. 1990;2:159. [PubMed: 2114928]
- 440.
- Masuda H, Ozols R F, Lai G M, Fojo A. et al. Increased DNA repair as a mechanism of acquired resistance to cis-diamminedichloroplatinum(II) in human ovarian cancer cell lines. Cancer Res. 1988;48:5713. [PubMed: 3139281]
- 441.
- Fisher R I, Erickson L C. 1-beta-D-arabinofuranosylcytosine and hydroxyurea production of cytotoxic synergy with cis-diamminedichloroplatinum(II) and modification of platinum-induced DNA interstrand cross-linking. Cancer Res. 1989;49:1383. [PubMed: 2924295]
- 442.
- Trujillo J M, Yang L Y. Synergism of 1-beta-D-arabinofuranosylcytosine and cis-diamminedichloroplatinum in their lethal efficacies against seven established cancer cell lines of gastrointestinal origin. Anticancer Res. 1989;9:197. [PubMed: 2705747]
- 443.
- Beck D J, Popoff S, Sancar A, Rupp W D. Reactions of the UVRABC excision nuclease with DNA damaged by diaminedichloroplatinum(II). Nucleic Acids Res. 1985;13:7395. [PMC free article: PMC322051] [PubMed: 3903663]
- 444.
- Husain I, Chaney S G, Sancar A. Repair of cis-platinum-DNA adducts by ABC excinuclease in vivo and in vitro. J Bacteriol. 1985;163:817. [PMC free article: PMC219204] [PubMed: 3897194]
- 445.
- Reardon J T, Vaisman A, Chaney S G, Sancar A. et al. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59:3968–3971. [PubMed: 10463593]
- 446.
- Dabholkar M, Vionnet J, Bostick-Bruton F, Yu J J, Reed E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest. 1994;94:703. [PMC free article: PMC296149] [PubMed: 8040325]
- 447.
- Li Q d, Tsang B, Bostick-Bruton F, Reed E. Modulation of excision repair cross complementation group 1 (ERCC-1) mRNA expression by pharmacological agents in human ovarian carcinoma cells. Biochem Pharmacol. 1999;57:347–353. [PubMed: 9933022]
- 448.
- Johnson S W, Perez R P, Godwin A K. et al. Role of platinum-DNA adduct formation and removal in cisplatin resistance in human ovarian cancer cell lines. Biochem Pharmacol. 1994;47:689. [PubMed: 8129746]
- 449.
- Bedford P, Fichtinger-Schepman A M, Shellard A A, Walker M C. et al. Differential repair of platinum-DNA adducts in human bladder and testicular tumor continuous cell lines. Cancer Res. 1988;48:3019. [PubMed: 3365691]
- 450.
- Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair [review] J Biol Chem. 1994;269:787. [PubMed: 8288625]
- 451.
- Chu G, Chang E. Cisplatin-resistant cells express increased levels of a factor that recognizes damaged DNA. Proc Natl Acad Sci U S A. 1990;87:3324. [PMC free article: PMC53892] [PubMed: 2333286]
- 452.
- Hwang B J, Chu G. Purification and characterization of a human protein that binds to damaged DNA. Biochemistry. 1993;32:1657. [PubMed: 8431446]
- 453.
- Huang J C, Zamble D B, Reardon J T, Lippard S J, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A. 1994;91:10394. [PMC free article: PMC45026] [PubMed: 7937961]
- 454.
- Lippard S J. Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science. 1992;256:234. [PubMed: 1566071]
- 455.
- Newman E M, Lu Y, Kashani-Sabet M, Kesavan V, Scanlon K J. Mechanisms of cross-resistance to methotrexate and 5-fluorouracil in an A2780 human ovarian carcinoma cell subline resistant to cisplatin. Biochem Pharmacol. 1988;37:443. [PubMed: 3337743]
- 456.
- Hill B T, Shellard S A, Hosking L K. et al. Characterization of a cisplatin-resistant human ovarian carcinoma cell line expressing cross-resistance to 5-fluorouracil but collateral sensitivity to methotrexate. Cancer Res. 1992;52:3110. [PubMed: 1591724]
- 457.
- Scanlon K J, Jiao L, Funato T. et al. Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein. Proc Natl Acad Sci U S A. 1991;88:10591–10595. [PMC free article: PMC52975] [PubMed: 1660142]
- 458.
- Scanlon K J, Kashani-Sabet M, Sowers L C. Overexpression of DNA replication and repair enzymes in cisplatin-resistant human colon carcinoma HCT8 cells and circumvention by azidothymidine. Cancer Commun. 1989;1:269. [PubMed: 2534792]
- 459.
- Antman K, Eder J P, Elias A. et al. High-dose combination alkylating agent preparative regimen with autologous bone marrow support: the Dana-Farber Cancer Institute/Beth Israel Hospital Experience. Cancer Treat Rep. 1987;71:119. [PubMed: 3542208]
- 460.
- van Besien K, Nichols C R, Tricot G. et al. Characteristics of engraftment after repeated autologous bone marrow transplantation. Exp Hematol. 1990;18:785. [PubMed: 2165911]
- 461.
- Friedman H S, Krischer J P, Burger P. et al. Treatment of children with progressive or recurrent brain tumors with carboplatin or iproplatin: a Pediatric Oncology Group randomized phase II study. J Clin Oncol. 1992;10:249. [PubMed: 1732426]
- 462.
- Lira-Puerto V, Silva A, Morris M. et al. Phase II trial of carboplatin or iproplatin in cervical cancer. Cancer Chemother Pharmacol. 1991;28:391. [PubMed: 1914084]
- 463.
- Nitschke R, Pratt C, Harris M. et al. Evaluation of CHIP (iproplatin) in recurrent pediatric malignant solid tumors. A phase II study. Invest New Drugs. 1992;10:93–96. [PubMed: 1323551]
- 464.
- Trask C, Silverstone A, Ash C M. et al. A randomized trial of carboplatin versus iproplatin in untreated advanced ovarian cancer. J Clin Oncol. 1991;9:1131. [PubMed: 2045855]
- 465.
- Vermorken J B, Gundersen S, Clavel M. et al. Randomized phase II trial of iproplatin and carboplatin in advanced breast cancer. Ann Oncol. 1993;4:303. [PubMed: 8518220]
- 466.
- Orourke T J, Weiss G R, New P. et al. Phase I clinical trial of ormaplatin (Tetraplatin, NSC 363812). Anticancer Drugs. 1994;5:520. [PubMed: 7858283]
- 467.
- Tutsch K d, Arzoomanian R z, Alberti D. et al. Phase I clinical and pharmacokinetic study of an one-hour infusion of ormaplatin (NSC 363812). Invest New Drugs. 1999;17:63–72. [PubMed: 10555124]
- 468.
- Extra J M, Espie M, Calvo F. et al. Phase I study of oxaliplatin in patients with advanced cancer. Cancer Chemother Pharmacol. 1990;25:299. [PubMed: 2295116]
- 469.
- Andre T, Louvet C, Raymond E. et al. Bimonthly high-dose leucovorin, 5-fluorouracil infusion and oxaliplatin (FOLFOX3) for metastatic colorectal cancer resistant to the same leucovorin and 5-fluorouracil regimen. Ann Oncol. 1998;9:1251–1253. [PubMed: 9862058]
- 470.
- Bleiberg H, de Gramont A. Oxaliplatin plus 5-fluorouracil: clinical experience in patients with advanced colorectal cancer. Semin Oncol. 1998;25:32–39. [PubMed: 9609106]
- 471.
- Wiseman L r. et al. Oxaliplatin—a review of its use in the management of metastatic colorectal cancer. Drugs Aging. 1999;14:459–475. [PubMed: 10408744]
- 472.
- Sessa C, Huinink W W T, du Bois A. Oxaliplatin in ovarian cancer. Ann Oncol. 1999;10:55–57. [PubMed: 10219454]
- 473.
- Soulie P, Bensmaine A, Garrino C. et al. Oxaliplatin/cisplatin (L-Ohp/Cddp) combination in heavily pretreated ovarian cancer. Eur J Cancer. 1997;33:1400–1406. [PubMed: 9337681]
- 474.
- Monnet I, Brienza S, Hugret F, et al. Phase II study of oxaliplatin in poor-prognosis non-small cell lung cancer (NSCLC). Item Corporate Author ATTIT, 1124. 34:1124-1127. [PubMed: 9849465]
- 475.
- Kelland L R, Abel G, MeKeage M J. et al. Preclinical antitumor evaluation of bis-acetato-ammine-dichloro-cyclohexylamine platinum (IV): an orally active platinum drug. Cancer Res. 1993;53:2581. [PubMed: 8388318]
- 476.
- Beale P, Raynaud F, Hanwell J. et al. Phase I study of oral JM216 given twice daily. Cancer Chemother Pharmacol. 1998;42:142–148. [PubMed: 9654114]
- 477.
- McKeage M J, Raynaud F, Ward J. et al. Phase I and pharmacokinetic study of an oral platinum complex given daily for 5 days in patients with cancer. J Clin Oncol. 1997;15:2691–2700. [PubMed: 9215842]
- 478.
- Sessa C, Minoia C, Ronchi A. et al. Phase I clinical and pharmacokinetic study of the oral platinum analogue JM216 given daily for 14 days. Ann Oncol. 1998;9:1315–1322. [PubMed: 9932162]
- 479.
- Parker R J, Vionet J A, Bostick-Bruton F, Reed E. Ormaplatin sensitivity/resistance in human ovarian cancer cells made resistant to cisplatin. Cancer Res. 1993;53:242. [PubMed: 8417816]
- 480.
- DeSimone P A, Yancey R S, Coupal J J. et al. Effect of a forced diuresis on the distribution and excretion (via urine and bile) of 195m platinum when given as 195m platinum cis-dichlorodiammineplatinum(II). Cancer Treat Rep. 1979;63:951. [PubMed: 466654]
- 481.
- Lange R C, Spencer R P, Harder H C. The antitumor agent cis-P1(NH3)2Cl2 distribution studies and dose calculations for 193m Pt. J Nucl Med. 1973;14:191. [PubMed: 4691408]
- 482.
- Goel R, Andrews P A, Pfeifle C E, Abramson I S, Kirmani S, Howell S B. Comparison of the pharmacokinetics of ultrafilterable cisplatin species detectable by derivatization with diethyldithiocarbamate or atomic absorption spectroscopy. Eur J Cancer. 1990;26:21. [PubMed: 2156545]
- 483.
- DeConti R C, Toftness B A U, Lange R C, Creasey W A. Clinical and pharmacological studies with cis-diamminedichloroplatinum(II). Cancer Res. 1973;33:1310. [PubMed: 4515709]
- 484.
- Repta AJ, Long DF. Cisplatin. Current Status and New Developments. Edited by AW Prestayko, ST Crooke, SK Carter. New York: Academic, 1980, p 285.
- 485.
- Patton TF, Repta AJ, Sternson LA. Clinical pharmacology of cisplatin. In Pharmacokinetics of Anticancer Agents in Humans. Edited by MM Ames, G Powis, JS Kovach. New York: Elsevier, 1983.
- 486.
- Reece P A, Bishop J F, Oliver I N. et al. Pharmacokinetics of unchanged carboplatin (CBDCA) in patients with small cell lung carcinoma. Cancer Chemother Pharmacol. 1987;19:326. [PubMed: 3036389]
- 487.
- Himmelstein K J, Patton T F, Belt R J, Taylor S, Repta A J, Sternson L A. Clinical kinetics on intact cisplatin and some related species. Clin Pharmacol Ther. 1981;29:658. [PubMed: 7194166]
- 488.
- Belt R J, Himmelstein K J, Patton T F. et al. Pharmacokinetics of non-protein-bound platinum species following administration of cis-dichlorodiamineplatinum(II). Cancer Treat Rep. 1979;63:1515. [PubMed: 498151]
- 489.
- Oguri S, Sakakibara T, Mase H. et al. Clinical pharmacokinetics of carboplatin. J Clin Pharmacol. 1988;28:208. [PubMed: 3283185]
- 490.
- Creaven P J, Pendylala L, Madajewicz S. Clinical development of iproplatin (CHIP). Drugs Exp Clin Res. 1986;12:287. [PubMed: 3732051]
- 491.
- Egorin M J, Jodrell D I. Utility of individualized carboplatin dosing alone and in combination regimens. Semin Oncol. 1992;19(1 Suppl 2):132. [PubMed: 1411624]
- 492.
- Calvert, H, Judson I, van der Vijgh W J. Platinum complexes in cancer medicine: pharmacokinetics and pharmacodynamics in relation to toxicity and therapeutic activity. Cancer Surv. 1993;17:189. [PubMed: 8137341]
- 493.
- Jodrell D I, Egorin M J, Canetta R M. et al. Relationships between carboplatin exposure and tumor response and toxicity in patients with ovarian cancer. J Clin Oncol. 1992;10:520. [PubMed: 1548516]
- 494.
- McLay E F, Howell S B. A review: intraperitoneal cisplatin in the management of patients with ovarian cancer. Gynecol Oncol. 1990;36:1. [PubMed: 2403957]
- 495.
- Speyer J L, Beller U, Colombo N. et al. Intraperitoneal carboplatin: favorable results in women with minimal residual ovarian cancer after cisplatin therapy. J Clin Oncol. 1990;8:1335. [PubMed: 2199620]
- 496.
- Speyer J L, Beller U, Colombo N. et al. Intraperitoneal carboplatin: favorable results in women with minimal residual ovarian cancer after cisplatin therapy. J Clin Oncol. 1990;8:1335. [PubMed: 2199620]
- 497.
- Howell S B. Intraperitoneal chemotherapy: the use of concurrent systemic neutralizing agents. Semin Oncol. 1985;12:17. [PubMed: 3876602]
- 498.
- Markman M, Cleary S, Howell S B. Nephrotoxicity of high-dose intracavitary cisplatin with intravenous thiosulfate protection. Eur J Cancer Clin Oncol. 1985;21:1015. [PubMed: 4065175]
- 499.
- Bacci G, Picci P, Ruggieri P. et al. Primary chemotherapy and delayed surgery (neoadjuvant chemotherapy) for osteosarcoma of the extremities. The Instituto Rissoli Experience in 127 patients treated preoperatively with intravenous methotrexate (high versus moderate doses) and intraarterial cisplatin. Cancer. 1990;65:2539. [PubMed: 2337871]
- 500.
- Calabro A, Singletary S E, Carrasco C H, Legha S S. Intra-arterial infusion chemotherapy in regionally advanced malignant melanoma. J Surg Oncol. 1990;43:239. [PubMed: 2325422]
- 501.
- Jaffe N, Raymond A K, Ayala A. et al. Effect of cumulative courses of intra-arterial cis-diamminedichloroplatin-II on the primary tumor in osteosarcoma. Cancer. 1989;63:63. [PubMed: 2910425]
- 502.
- Kashdan B J, Sullivan K L, Lackman R D. et al. Extremity osteosarcomas: intra-arterial chemotherapy and limb-sparing resection with 2-year follow-up. Radiology. 1990;177:95. [PubMed: 2144653]
- 503.
- Follezou J Y, Fauchon F, Chiras J. Intra-arterial infusion of carboplatin in the treatment of malignant gliomas: a phase II study. Neoplasma. 1989;36:349. [PubMed: 2544815]
- 504.
- Lehane D E, Bryan R N, Horowitz B. et al. Intra-arterial cis-platinum chemotherapy for patients with primary and metastatic brain tumors. Cancer Drug Deliv. 1983;1:69. [PubMed: 6544119]
- 505.
- Stewart D J, Grahovac Z, Hugenholtz H. et al. Combined intra-arterial and systemic chemotherapy for intracerebral tumors. Neurosurgery. 1987;21:207. [PubMed: 2443873]
- 506.
- Baker S R, Wheeler R. Intraarterial chemotherapy for head and neck cancer. Part 2: clinical experience. Head Neck Surg. 1984;6:751. [PubMed: 6198307]
- 507.
- Mortimer J E, Taylor M E, Schulman S, Cummings C, Weymuller E Jr, Laramore G. Feasibility and efficacy of weekly intra-arterial cisplatin in locally advanced (stage III and IV) head and neck cancers. J Clin Oncol. 1988;6:969. [PubMed: 3373266]
- 508.
- Kasugai H, Kojima J, Tatsuta M. et al. Treatment of hepatocellular carcinoma by transcatheter arterial cisplatin and ethiodized oil. Gastroenterology. 1989;97:965. [PubMed: 2550311]
- 509.
- Eapen L, Stewart D, Danjoux C. et al. Intraarterial cisplatin and concurrent radiation for locally advanced bladder cancer. J Clin Oncol. 1989;7:230. [PubMed: 2915239]
- 510.
- Jacobs S C, Menashe D S. Intra-arterial chemotherapy for bladder cancer. Prog Clin Biol Res. 1990;350:101. [PubMed: 1696738]
- 511.
- Blumenreich M S, Needles B, Yagoda A. et al. Intravesical cisplatin for superficial bladder tumors. Cancer. 1982;50:863. [PubMed: 7201343]
- 512.
- Horn Y, Eidelman A, Walach N, Waron M, Barak F. Intravesical chemotherapy of superficial bladder tumors in a controlled trial with cis-platinum versus cis-platinum plus hyaluronidase. J Surg Oncol. 1985;28:304. [PubMed: 3884905]
- 513.
- Liopis B, Gallego J, Mompo J A. et al. Thiotepa versus adriamycin versus cis-platinum in the intravesical prophylaxis of superficial bladder tumors. Eur Urol. 1985;11:73. [PubMed: 3924624]
- 514.
- Fiorentino M V, Daniele O, Morandi P. et al. Intrapericardial instillation of cis-platin in malignant pericardial effusion. Cancer. 1988;62:1904. [PubMed: 3167805]
- 515.
- Markman M, Howell S B. Intrapericardial instillation of cisplatin in a patient with a large malignant effusion. Cancer Drug Deliv. 1985;2:49. [PubMed: 4052925]
- 516.
- Gottlieb J A, Drewinko B. Review of the current clinical status of platinum coordination complexes in cancer chemotherapy. Cancer Chemother Rep. 1975;59:621. [PubMed: 1106840]
- 517.
- Piel I J, Perlia C P. Phase II study of cis-dichlorodiammine platinum (ll) (NSC-119875) in combination with cyclophosphamide (NSC-26271) in the treatment of human malignancies. Cancer Chemother Rep. 1975;59:995. [PubMed: 1106849]
- 518.
- Dentino M, Luft F C, Yum M N. et al. Long term effect of cis-diamminedichloride platinum (CDDP) on renal function and structure in man. Cancer. 1978;41:1274. [PubMed: 638991]
- 519.
- Schilsky R L, Anderson T. Hypomagnesemia and renal magnesium wasting in patients receiving cisplatin. Ann Intern Med. 1979;90:929. [PubMed: 375794]
- 520.
- Gonzales-Vitale J C, Hayes D M, Cvitkovic E, Sternberg S S. The renal pathology in clinical trials of cis-platinum (ll) diamminedichloride. Cancer. 1977;39:1362. [PubMed: 851939]
- 521.
- Madias N E, Harrington J T. Platinum nephrotoxicity. Am J Med. 1978;65:307. [PubMed: 99034]
- 522.
- Chary K K, Higby D J, Henderson E S, Swinerton K D. Phase I study of high-dose cis-dichlorodiammineplatinum(II) with forced diuresis. Cancer Treat Rep. 1977;61:367. [PubMed: 266972]
- 523.
- Hayes D M, Cvitkovic E, Golbey R B. et al. High dose cis-platinum diammine dichloride: amelioration of renal toxicity by mannitol diuresis. Cancer. 1977;39:1372. [PubMed: 856437]
- 524.
- Berry J M, Jacobs C, Sikic B. et al. Modification of cisplatin toxicity with diethyldithiocarbamate. J Clin Oncol. 1990;8:1585. [PubMed: 2167955]
- 525.
- Skinner R, Pearson A D, Amineddine H A. et al. Ototoxicity of cisplatinum in children and adolescents. Br J Cancer. 1990;61:927. [PMC free article: PMC1971678] [PubMed: 2372498]
- 526.
- Kretschmar C S, Warren M P, Lavally B L. et al. Ototoxicity of preradiation cisplatin for children with central nervous system tumors. J Clin Oncol. 1990;8:191. [PubMed: 2358836]
- 527.
- Black F O, Myers E N, Schramm V L. et al. Cisplatin vestibular ototoxicity: preliminary report. Laryngoscope. 1982;92:1363. [PubMed: 6983636]
- 528.
- Wright C G, Schaefer S D. Inner ear histopathology in patients treated with cis-platinum. Laryngoscope. 1982;92:1408. [PubMed: 6983637]
- 529.
- Hadjilaskari P, Fengler R, Hartmann R, Henze G. Ototoxicity of cisplatin in children with malignant diseases. Klin Padiatr. 1989;201:316. [PubMed: 2779137]
- 530.
- Vermorken J B, Kapteijn T S, Hart A A, Pinedo H M. Ototoxicity of cis-diamminedichloroplatinum (ll): influence of dose, schedule and mode of administration. Eur J Cancer Clin Oncol. 1983;19:53. [PubMed: 6682776]
- 531.
- Granowetter L, Rosenstock J G, Packer R J. Enhanced cis-platinum neurotoxicity in pediatric patients with brain tumors. J Neurooncol. 1983;1:293. [PubMed: 6687235]
- 532.
- Walker D A, Pillow J, Waters K D, Keir E. Enhanced cis-platinum ototoxicity in children with brain tumours who have received simultaneous or prior cranial irradiation. Med Pediatr Oncol. 1989;17:48. [PubMed: 2913475]
- 533.
- Boheim K, Bichler E. Cisplatin-induced ototoxicity: audiometric findings and experimental cochlear pathology. Arch Otorhinolaryngol. 1985;242:1. [PubMed: 4041173]
- 534.
- Kohn S, Fradis M, Pratt H. et al. Cisplatin ototoxicity in guinea pigs with special reference to toxic effects in the stria vascularis. Laryngoscope. 1988;98 (8 Pt 1):865. [PubMed: 3398664]
- 535.
- Moroso M J, Blair R L. A review of cis-platinum ototoxicity. J Otolaryngol. 1983;12:365. [PubMed: 6686617]
- 536.
- Strauss M, Towfighi J, Lord S. et al. Cis-platinum ototoxicity: clinical experience and temporal bone histopathology. Laryngoscope. 1983;93:1554. [PubMed: 6685804]
- 537.
- Anniko M, Sobin A. Cisplatin: evaluation of its ototoxic potential. Am J Otolaryngol. 1986;7:276. [PubMed: 3752388]
- 538.
- Von Hoff D D, Schilsky R, Reichert C M. et al. Toxic effects of cis-dichlorodiammineplatinum(II) in man. Cancer Treat Rep. 1979;63:1527. [PubMed: 387223]
- 539.
- Bruck W, Heise E, Friede R L. Leukoencephalopathy after cisplatin therapy. Clin Neuropathol. 1989;8:263. [PubMed: 2620478]
- 540.
- Cattaneo M T, Filipazzi V, Piazza E. et al. Transient blindness and seizure associated with cisplatin therapy. J Cancer Res Clin Oncol. 1988;114:528. [PubMed: 3182914]
- 541.
- Frustaci S, Barzan L, Comoretto R. et al. Local neurotoxicity after intra-arterial cisplatin in head and neck cancer. Cancer Treat Rep. 1987;71:257. [PubMed: 3815392]
- 542.
- Pomes A, Frustaci S, Cattaino G. et al. Local neurotoxicity of cisplatin after intra-arterial chemotherapy. Acta Neurol Scand. 1986;73:302. [PubMed: 3716771]
- 543.
- Hansen S W, Helweg-Larsen S, Trojaborg W. Long-term neurotoxicity in patients treated with cisplatin, vinblastine, and bleomycin for metastatic germ cell cancer. J Clin Oncol. 1989;7:1457. [PubMed: 2476531]
- 544.
- Elderson A, Gerritsen van der Hoop R. et al. Vibration perception and thermoperception as quantitative measurements in the monitoring of cisplatin induced neurotoxicity. J Neurol Sci. 1989;93:167. [PubMed: 2592981]
- 545.
- Busse O, Aigner K, Wilimzig H. Peripheral nerve damage following isolated extremity perfusion with cis-platinum. Recent Results Cancer Res. 1983;86:264. [PubMed: 6316430]
- 546.
- Neuwelt E A, Glasberg M, Frenkel E, Barnett P. Neurotoxicity of chemotherapeutic agents after blood-brain barrier modification: neuropathological studies. Ann Neurol. 1983;14:316. [PubMed: 6195955]
- 547.
- Mahaley M S Jr, Hipp S W, Dropcho E J. et al. Intracarotid cisplatin chemotherapy for recurrent gliomas. J Neurosurg. 1989;70:371. [PubMed: 2536804]
- 548.
- Pratt C B, Goren M P, Meyer W H. et al. Ifosfamide neurotoxicity is related to previous cisplatin treatment for pediatric solid tumors. J Clin Oncol. 1990;8:1399. [PubMed: 2380760]
- 549.
- Ozols R F. Cisplatin dose intensity. Semin Oncol. 1989;16:22. [PubMed: 2669135]
- 550.
- van der Hoop R G, Hamers F P, Neijt J P. et al. Protection against cisplatin induced neurotoxicity by ORG 2766: histological and electrophysiological evidence. J Neurol Sci. 1994;126:109. [PubMed: 7853014]
- 551.
- van der Hoop R G, Vecht C J, van der Burg M E. et al. Prevention of cisplatin neurotoxicity with an ACTH(4-9) analogue in patients with ovarian cancer. N Engl J Med. 1990;322:89. [PubMed: 2152972]
- 552.
- Nichols C R, Tricot G, Williams S D. et al. Dose-intensive chemotherapy in refractory germ cell cancerùa phase I/II trial of high-dose carboplatin and etoposide with autologous bone marrow transplantation. J Clin Oncol. 1989;7:932. [PubMed: 2544687]
- 553.
- Paolozzi F P, Gaver R, Poiesz B J. et al. Phase I-preliminary phase II trial of iproplatin, a cisplatin analogue. Invest New Drugs. 1988;6:199. [PubMed: 3192385]
- 554.
- van Zandwijk N, Ten Bokkel Huinink W W, Wanders J. et al. Dose-finding studies with carboplatin, ifosfamide, etoposide, and mesna in non-small cell lung cancer. Semin Oncol. 1990;17:16. [PubMed: 2154854]
- 555.
- Prestayko AW. Cisplatin and analogues: a new class of anticancer drugs. In Cancer and Chemotherapy, vol III, Antineoplastic Agents. Edited by ST Crooke, AW Prestayko. New York: Academic, 1981, p 133.
- 556.
- Hawthorn J, Ostler K J, Andrews P L. The role of the abdominal visceral innervation and 5-hydroxytryptamine M-receptors in vomiting induced by the cytotoxic drugs cyclophosphamide and cis-platin in the ferret. Q J Exp Physiol. 1988;73:7. [PubMed: 3347698]
- 557.
- Higgins G A, Kilpatrick G J, Bunce K T. et al. 5-HT3 receptor antagonists injected into the area postrema inhibit cisplatin-induced emesis in the ferret. Br J Pharmacol. 1989;97:247. [PMC free article: PMC1854462] [PubMed: 2720310]
- 558.
- McCarthy L E, Borison H L. Cisplatin-induced vomiting eliminated by ablation of the area postrema in cats. Cancer Treat Rep. 1984;68:401. [PubMed: 6538113]
- 559.
- Gralla R J, Itri L M, Pisko S E. et al. Antiemetic efficacy of high-dose metoclopramide: randomized trials with placebo and prochlorperazine in patients with chemotherapy-induced nausea and vomiting. N Engl J Med. 1981;305:905. [PubMed: 7024807]
- 560.
- Kahn T, Elias E G, Mason G R. A single dose of metoclopramide in the control of vomiting from cis-dichlorodiammineplatinum(II) in man. Cancer Treat Rep. 1978;62:1106. [PubMed: 688250]
- 561.
- Aapro M S, Plezia P M, Alberts D S. et al. Double-blind crossover study of the antiemetic efficacy of high-dose dexamethasone versus high-dose metoclopramide. J Clin Oncol. 1984;2:466. [PubMed: 6539363]
- 562.
- Benrubi G I, Norvell M, Nuss R C, Robinson H. The use of methylprednisolone and metoclopramide in control of emesis in patients receiving cis-platinum. Gynecol Oncol. 1985;21:306. [PubMed: 4040050]
- 563.
- Gez E, Ben Yosef R, Catane R. et al. Chlorpromazine and dexamethasone versus high-dose metoclopramide and dexamethasone in patients receiving cancer chemotherapy, particularly cis-platinum: a prospective randomized crossover study. Oncology. 1989;46:150. [PubMed: 2654792]
- 564.
- Arseneau J, Blessing J A, Stehman F B, McGehee R. A phase II study of carboplatin in advanced squamous cell carcinoma of the cervix (a Gynecologic Oncology Group Study). Invest New Drugs. 1986;4:187. [PubMed: 3525449]
- 565.
- Eisenberger M, Hornedo J, Silva H. et al. Carboplatin (NSC-241–240): an active platinum analog for the treatment of squamous-cell carcinoma of the head and neck. J Clin Oncol. 1986;4:1506. [PubMed: 3531424]
- 566.
- Goldenberg A S, Kelsen D, Dougherty J, Magill G. Phase II study of CHIP chemotherapy in advanced adenocarcinomas of the upper gastrointestinal tract. Invest New Drugs. 1990;8:71. [PubMed: 2345073]
- 567.
- Reed E, Kohn KW. Platinum analogues. In Cancer Chemotherapy: Principles and Practice. Edited by BA Chabner, JM Collins. Philadelphia: Lippincott, 1990, p 475.
- 568.
- Kleinerman ES, Zwelling LA. The effect of cis-diamminedichloroplatinum(II) on immune function in vitro and in vivo.
- 569.
- Onsrud M, Bosnes V, Graham I. Cis-Platinum as adjunctive to surgery in early stage ovarian carcinoma: effects on lymphoid cell subpopulations. Gynecol Oncol. 1986;23:323. [PubMed: 3957118]
- Alkylating Agents and Platinum Antitumor Compounds - Holland-Frei Cancer Medicin...Alkylating Agents and Platinum Antitumor Compounds - Holland-Frei Cancer Medicine
Your browsing activity is empty.
Activity recording is turned off.
See more...