

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.

Proteins Derived from gag
The Gag protein is the precursor to the internal structural protein of all retroviruses. That the internal structural proteins of retroviruses are derived from a single polypeptide was first recognized in ASLV, when antibodies were used to identify newly synthesized radioactively labeled viral proteins (Vogt and Eisenman 1973). Soon afterward, the same conclusion was drawn for MLV (Jamjoon et al. 1975; van Zaane et al. 1975). Expression of gag alone leads to assembly of immature virus-like particles that bud from the plasma membrane (Chapter 7). These particles lack the surface projections comprising the Env proteins, but they are otherwise indistinguishable in thin-section electron micrographs from virions formed by complete genomes with an inactivating mutation in protease. In virion assembly, Gag proteins must interact with each other, with components in the plasma membrane, with the genomic RNA, and probably with Env proteins and with cellular proteins as well. Fundamental to understanding the function of Gag is the fact that this protein is organized into regions, which are proteolytically liberated as the separate mature Gag proteins during viral maturation (Fig. 7). Because proteolytic cleavage occurs late in assembly, during or after the last stages of budding, virions contain equimolar mixtures of the mature proteins. According to older reports, there are approximately 2000 molecules of each Gag protein in a virion, which together represent about three quarters of the protein component of the virus. Although this is a conventionally accepted number, the exact number of copies of Gag in a virion has not been established. In fact, the population of particles may not be completely uniform in size. Neither thin-section electron microscopy nor rate zonal sedimentation is sufficiently precise to address this question, and available cryo-EM measurements show a spread in diameter of MLV particles and HIV-like particles (see Fig. 2) (E. Kubalek et al.; S.D. Fuller et al.; both in prep.).

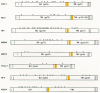
All Gag proteins are organized in the same order from the amino terminus to the carboxyl terminus, with domains that are cleaved into the following proteins: (NH2)-MA-X-CA-NC-Y-(COOH) (for review, see Wills and Craven 1991). X and Y represent segments that each may be cleaved into one or more small proteins or peptides or may be absent altogether. Thus, the “minimal” Gag protein is the unit MA-CA-NC. Examples of the structural organization of Gag proteins of prototypic retroviruses are given in Figure 7.
MA Protein
In all retroviruses, the amino-terminal domain of Gag gives rise to the MA protein (membrane-associated, or matrix). Both in ASLV and in MLV, this protein can be crosslinked to radioactively labeled phospholipid (phosphatidylethanolamine, PE) when virions are treated with amino group-reactive bifunctional reagents ( Pepinsky and Vogt 1979). Under similar conditions, matrix (M) proteins of influenza virus and vesicular stomatitis virus also became crosslinked to lipid. The sites of crosslinking of the MA proteins of ASLV and MLV are in the amino-terminal several dozen amino acid residues of these proteins (Pepinsky and Vogt 1984). The lipid crosslinking implies proximity between lysine residues on the protein and the amino group of PE, but it cannot be taken as evidence that MA actually interdigitates with the lipid bilayer. Early experiments with surface-specific iodination of MLV suggested that some MA molecules might be exposed on the outside of the virion (Barbacid and Aaronson 1978). Consistent with this idea, in MLV (Schiff-Maker and Rosenberg 1986), the plasma membrane of infected cells shows some reactivity with anti-Gag monoclonal antibodies. In addition, in HIV, anti-MA monoclonal antibodies have been reported to react with the surface of virions as well as to neutralize the virus (Papsidero et al. 1989). The authenticity and significance of such surface-localized Gag molecules are not clear. An interpretation that has not been rigorously excluded is that in the virion, a small fraction of Gag molecules actually traverses the membrane. In considering possible cell surface localization of MLV Gag proteins, it is important to keep in mind that a fraction of the Gag translation products (of viruses in this genus only) are initiated at an upstream site, glycosylated, and exported from the cell in an uncleaved form (Edwards and Fan 1979, 1980). Although these products are not incorporated into virions, they can confuse attempts to localize Gag proteins in the cell. In any case, by most biochemical criteria, retroviral MA proteins appear to be peripheral membrane proteins, not integral membrane proteins, consistent with the absence of long stretches of hydrophobic amino acids like those typically found spanning a membrane.
The finding that most Gag proteins are modified by myristylation at their amino termini provided a major clue to MA function (Henderson et al. 1983). Myristate is a 14-carbon fatty acid that is added cotranslationally to many cellular proteins associated with membranes and also to some proteins that remain cytosolic (for review, see Grand 1989; Schmidt 1989). The consensus sequence for myristylation is Met-Gly-X-X..Ser/Thr. After the initiating methionyl residue is removed, the fatty acid is linked via an amide bond to the free amino group of the glycyl residue. Mutagenesis was used to show, first for MLV (Rein et al. 1986), that myristylation is essential for retroviral assembly. Alteration of the glycine residue leads to a block in the budding of particles and an accumulation of Gag inside the cell (Chapter 7). The same phenotype is observed in HIV (Göttlinger et al. 1989; Bryant and Ratner 1990). Prevention of myristylation of the M-PMV Gag protein does not prevent formation of the immature particles in the cytoplasm, but rather prevents their transport to, or stable association with, the plasma membrane (Rhee and Hunter 1987).
Several arguments suggest that a myristate moiety is not the only means of targeting Gag to the membrane. The Gag proteins of some viruses, such as ASLV and EIAV, are not myristylated. The amino-terminal half of avian MA has been mapped as the part of this protein that is essential for membrane binding. Deletions in this region prevent membrane binding and thus virion assembly, but this function can be replaced by a short amino-terminal sequence from the Src oncoprotein. This small fragment of Src is myristylated, but not all myristylated peptides function to rescue membrane binding in experiments of this type. Although ASLV MA is not myristylated, it is modified in two other ways. The initiating amino-terminal methionine is acetylated (Palmiter et al. 1978), and about one half of the molecules in virions are phosphorylated (Lai 1976). The single major phosphorylation site can be mutated without loss of infectivity of the virus (J.W. Wills, pers. comm.). The functions of these modifications remain unknown.
Myristylation is not the only feature of MA necessary for association with the membrane. Biochemical evidence suggests that a segment of the HIV-1 MA polypeptide with multiple positive charges, somewhat downstream from the amino terminus, interacts with acidic phospholipids in vitro (Zhou et al. 1994). Gag protein made by in vitro translation binds to a crude mixture of membranous vesicles from lysed cells. Efficient binding is dependent both on the fatty acid modification at the amino-terminal glycine residue and on the basic sequences farther downstream. Many, but not all, MA proteins also have clusters of basic residues in this region. A major interaction of MA proteins and the virion lipid bilayer may thus involve ionic interactions of this type, which would be consistent with the localization of lipid-protein crosslinks in the amino-terminal regions of several viruses. Since the mixture of lipids and chlolesterol is not identical in virions and the plasma membrane, the affinity of the MA domain of the Gag polyprotein for different lipids may not be the same. It is also possible that the virus chooses specialized areas of the plasma membrane that are already enriched in certain lipids for budding, perhaps because they are free of cortical cytoskeletal elements. In vitro assembly systems may be able to help address these issues.
MA may also interact with the Env proteins during budding. Portions of MA can be deleted from ASLV and HIV-1 Gag proteins without impairing its ability to assemble and bud from the membrane (Chapter 7). Some of these deletions prevent incorporation of HIV envelope glycoproteins into virions (Yu et al. 1992; Fäcke et al. 1993), suggesting that at least a part of the MA domain forms a contact with the cytoplasmic portion of Env. Such contacts are also indicated by the reported ability of ASLV MA to be crosslinked to Env with bifunctional reagents (Gebhardt et al. 1984) and by mutations in M-PMV MA that suppress a mutation in the carboxy-terminal domain of Env (Brody et al. 1992). Finally, chimeric Gag proteins containing MA from HIV-1 and the remainder from visna virus specifically incorporate the HIV Env protein into budding virions (Dorfman et al. 1994a).
In seeming contradiction to the need for specific MA-Env interaction, most or all of the cytoplasmic tail of Env of HIV-1 and ASLV can be deleted without loss of incorporation of the protein into virions and without loss of infectivity (Perez et al. 1987; Wilk et al. 1992). Similarly, retroviruses are well known for their ability to incorporate into infectious virions (called pseudotypes) envelope proteins from retroviruses in other genera and even from other viral families. The best studied pseudotypes are those with the vesicular stomatitis virus (VSV) G protein (Zavada 1972; Huang et al. 1973; Love and Weiss 1974; Emi et al. 1991). An ASLV genome has been engineered to carry the gene for influenza virus hemagglutinin in place of env, and the resulting virus is infectious at a low level (Dong et al. 1992). Since the carboxy-terminal domains of these diverse transmembrane proteins show no obvious sequence similarities, it is unclear what mechanisms select them for inclusion into a virion. The same question underlies the preferential incorporation of certain host-cell membrane proteins into virions. One speculation is that incorporation of plasma membrane proteins into a budding virion occurs when they are highly mobile and not tethered to the cytoskeleton as are many cellular membrane proteins (Chapter 7).
The three-dimensional structure of HIV-1 MA protein in solution has been deduced by nuclear magnetic resonance (NMR) (Matthews et al. 1994; Massiah et al. 1994), a powerful technique used to study small proteins that are highly soluble. In solution, MA is a monomer consisting of five helices joined by short loops or β-stands, four of the helices surrounding a fifth to form a hydrophobic core (Fig. 9A). In this structure, most of the basic residues, including those near the amino terminus, are positioned at one end of the molecule. The NMR structures have been confirmed and extended by X-ray crystallography (Hill et al. 1996) for both HIV-1 and the related simian immunodeficiency virus (SIV) MA protein (Rao et al. 1995). Both of these lentiviral MA proteins are trimeric in the crystal, with a globular amino-terminal domain and a smaller carboxy-terminal domain projecting away. The strongly positively charged regions on one side of each of the globular domains come together to form a kind of platform. The amino termini of the three subunits, which would be myristylated in the virus, are positioned in the same region. The shape of the trimer is thus consistent with the notion that the myristates insert into the hydrophobic portion of the membrane and the basic charges interact with the phosphate of the lipid head groups (Fig. 9B). Perhaps the cytoplasmic tail of the multimeric Env transmembrane complex is accommodated in the interstices between trimers. Although the three-dimensional structure of MA is very useful as a guide to understanding virion structure, it should be pointed out that the trimeric nature of the protein in crystals does not necessarily imply that this is the form in the virion. Furthermore, since all of the structural determinations to date have been carried out on unmyristylated protein, it is not excluded that this fatty acid modification alters the folding of the polypeptide in vivo. In addition, biochemical evidence suggests that the conformation of HIV MA as a mature protein is not identical with that of the MA domain as part of Gag (Zhou and Resh 1996).
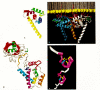
MA proteins of at least some viruses can bind RNA in vitro. The possible role of RNA binding in the viral life cycle is unclear and may depend on the virus in question. ASLV MA binds to RNA in a sequence-independent fashion (Steeg and Vogt 1990), although early reports claimed that this binding is specific for viral RNA. In contrast, one form of the MA protein from BLV apparently can recognize RNAs carrying sequences near the 5′end of the genomic RNA (Katoh et al. 1991), fueling the speculation that this protein domain has a role in packaging of RNA into virions. However, deletion analyses using ASLV suggest that MA does not have a role in RNA packaging (Chapter 7). In HIV-1, MA has been found to accompany the newly synthesized viral DNA into the nucleus (Bukrinsky et al. 1993a) and, along with Vpr (see below), may be a factor that directs migration of the preintegration complex. There is a nuclear localization signal in HIV MA (Bukrinsky et al. 1993b), mapping to the same highly basic stretch of amino acids near the amino terminus, that has been inferred to be important for membrane interaction (Zhou et al. 1994). The phenotypes of mutant HIV strains constructed with alterations in this sequence suggest that the nuclear targeting function may be important for the ability of HIV to infect some types of resting cells (von Schwedler et al. 1994). Tyrosine phosphorylation on a minor fraction of HIV-1 MA molecules is reported to be a regulatory signal for directing MA to the nucleus instead of the infected cell membrane (Gallay et al. 1995a), apparently inducing binding of MA molecules to the integrase (Gallay et al. 1995b).
In two cases, a domain of Gag corresponding approximately to MA has been found to be capable of budding from the membrane, in the absence of any capsid or nucleocapsid sequences (Chapter 6). In ASLV, the first 180 amino acid residues of Gag, which include MA and extend into the amino-terminal region of p10, can form a membrane-enclosed particle (see Weldon and Wills 1993). However, the density of this particle is much less than that of wild-type virus. SIV MA expressed by itself is also released from cells as a particle, with the dimensions but not the morphology of bona fide virions (Gonzalez et al. 1993). These observations imply that MA is capable of membrane interactions as well as protein-protein interactions without help from the adjoining Gag sequences. Presumably, these functions reflect part of the role that the MA domain plays in the wild-type virus.
CA Protein
Of all the Gag proteins, the capsid (CA) is the easiest to recognize on SDS-polyacrylamide gel electrophoresis because it is the largest, approximately 200–270 amino acid residues in size. It is typically very antigenic; polyclonal antisera to other Gag proteins purified from virions usually also react with CA, due to low levels of contamination of the purified immunogen by CA. The primary amino acid sequences of CA proteins from most retroviruses, except the spumaviruses, show similarity in the 20-amino-acid-residue long “major homology region” or MHR. This segment of CA is the most highly conserved sequence in the Gag protein, along with the Cys-His motif in the nucleocapsid, which also is absent in spumaviruses. CA can be purified readily as a soluble protein both from viral particles and from recombinant-DNA-based expression systems. The three-dimensional folding of the HIV-1 CA polypeptide is being unraveled and may provide clues to its functions. A solution structure (Gitti et al. 1996) and a crystal structure (Gamble et al. 1996) of the fragment corresponding to the amino-terminal 151 amino acid residues of the protein have been determined (Fig. 9C). The latter includes the cellular protein cyclophilin A as it is bound to CA. In addition, the structure of the intact protein complexed with a monoclonal antibody fragment has been solved in part (Momany et al. 1996). Unlike the majority of known CA proteins from diverse icosahedral viruses, which show a similar folding characterized by eight β strands, CA is largely helical, as predicted earlier from biochemical data (Burns et al. 1990; Ehrlich et al. 1994). The helices in the CA fragment pack together to form an arrowhead shape, with the carboxy-terminal end at the tip and the amino-terminal end near the base. The amino terminus itself is folded back so that it is buried in the molecule. This implies a conformational change upon cleavage by PR, since the MA-CA junction must be stretched out to be readily accessible to viral protease.
The exact structural function of CA in the mature viral particle has not been elucidated, but the protein is believed to form a shell surrounding the ribonucleoprotein complex that contains the genomic RNA, as originally suggested in the 1970s. The possibility that other Gag proteins, perhaps in lower amounts than CA, also form part of the shell has not been ruled out. This shell is most appropriately referred to as the “capsid.” The capsid together with the components it encloses are then referred to as the “core.” These two terms frequently have been used interchangeably, but such usage promotes confusion between the proteins that form the shell and the proteins and RNA inside the shell. As discussed above, by thin-section electron microscopy, cores can be approximately spherical or bar- or cone-shaped, depending on the virus. Presumably, the nature of CA determines the shape of the core, but this supposition has not been tested critically. Since the CA and nucleocapsid domains in Gag seem to function in consort as a unit in assembly (Chapter 7), the nucleocapsid could also play a part in the shape of the core.
Cores can be isolated from mature viral particles treated with nonionic detergents, but usually only in poor yield and in impure form. Apparently, they are fragile, and the detergents used to strip lipid and the MA layer under the lipid membrane typically result in their disintegration. This fragility may reflect a need for the core to dissociate readily from the membrane soon after entry into a newly infected cell. Nevertheless, there are several older reports on methods of core isolation and biochemical activities of cores (Stromberg et al. 1974; Durbin and Manning 1982). Interpretations of some of the published data, for example, the numerous early reports in which detergent-treated viral particles were used in reverse transcription assays, are difficult due to the absence of electron microscope data. The cores of EIAV may be less labile than other retroviral cores, making this retrovirus a promising model system to study core structure and function (Roberts and Oroszlan 1989).
The biological function of the capsid shell is not known. Other prototypic enveloped RNA viruses, such as VSV (a rhabdovirus), Sendai virus (a paramyxovirus), hepatitis B virus (a hepadnavirus), influenza virus (a myxovirus), and Sindbis virus (an alphavirus), do not have such a separate shell between the viral membrane and its associated matrix protein, and the RNA genome and its associated nucleocapsid protein. In all of these viruses, the nucleocapsid proteins are much larger than retroviral NC proteins, and thus perhaps carry out the functions of CA and NC together. In several of these cases, for example, the alphaviruses (Choi et al. 1991), the segment of polypeptide known to bind to RNA forms a separate domain. The same holds for the coat proteins of some nonenveloped plant viruses. For example, the cowpea chlorotic virus capsid protein has an amino-terminal RNA-binding domain that is distinct from the domain mediating protein-protein interactions (Zhao et al. 1995). The proteolytic cleavage event that severs the connection between the retroviral NC and CA, which is probably involved directly in morphological maturation of the virus, may be necessary for a subsequent step in the infectious cycle, for example, proper disassembly upon infection.
Several lines of evidence suggest that CA is important early in infection. One is the phenomenon of Fv1 restriction (Jolicoeur 1979). In MLV, a single-amino-acid residue in CA determines the N- or B-cell tropism of the virus. B-tropic MLV efficiently infects BALB/c cells but has only limited ability to replicate in NIH-3T3 cells, showing about a 100-fold lower titer. The opposite is true for N-tropic virus. The Fv1 locus in the host determines whether a cell will be of the B- or N-type. The protein product of this gene is presumed to interact with CA in the infecting virus. Fv1 restriction results in a defect prior to integration of the newly made DNA copy of the infecting virus (Chapter 5). The Fv1 gene has recently been cloned, but its function remains to be elucidated (Best et al. 1996). Conclusions from the Fv1 data are reinforced by the report that the MLV CA protein is found in the preintegration complex in the newly infected cell (Bowerman et al. 1989). A second line of evidence for an important early role for CA is based on the requirement for cyclophilin A binding for assembly of an infectious HIV-1 virion (see below). Viral particles shed from cells treated with cyclosporin, a drug that prevents incorporation of cyclophilin A into virions, are blocked in replication at a step subsequent to entry but prior to reverse transcription (Braaten et al. 1996). Cyclophilin A interacts with a loop containing proline 90 in an unusual trans-conformation. According to one hypothesis (Gamble et al. 1996), the biological function of cyclophilin A binding to CA might be to destabilize the shell of CA proteins, thereby facilitating disassembly upon infection.
Numerous molecular genetic analyses have been carried out to study the function of CA in assembly, largely in the ASLV and HIV systems (Chapter 7). Data suggest that the amino-terminal portion of the HIV protein is less important for budding of a virus-like particle than the carboxy-terminal portion (Dorfman et al. 1994b). Given its conservation in evolution, the MHR sequence might have a critical role in the function of CA. To date, mutational analyses of the MHR region of CA have not provided an altogether consistent phenotype, although a number of different mutations do compromise assembly (Strambio de Castillia and Hunter 1992; Mammano et al. 1994; Craven et al. 1995). Surprisingly, large portions of the ASLV CA, extending in the aggregate over most or all of this domain in Gag, can be deleted without loss of budding (Wills and Craven 1991). This implies that the CA shell is not required for formation of at least some type of enveloped particle, at least for ASLV. In mutants with small deletions in CA, the diameter of the virus-like particle is larger than wild type or is heterogeneous, or the morphology of the particle is altered (N.K. Krishna et al., in prep.). The behavior of some CA mutants suggests that the CA domain has an important role in core stability; however, more indirect effects are difficult to rule out. When wild-type ASLV virions are treated with nonionic detergent under defined conditions, a proportion of the CA protein is found in a form that can be collected by centrifugation. In mutant particles with partial deletions of CA, this protein is completely solubilized by detergent (Craven et al. 1995). The same effect also is seen for deletions in the spacer peptide between CA and the nucleocapsid (see below).
The biochemical properties of the mature Gag proteins as isolated entities should give information about their function in vivo. At high concentration, HIV-1 CA purified from E. coli strains containing appropriate expression vectors can assemble into regular fibrous structures in vitro (Ehrlich et al. 1992). The protein-protein interactions in these structures are likely to be similar to those in genuine virions. Both for HIV-1 and for ASLV, the CA-NC protein (two linked domains as they are found in the Gag protein before cleavage) also has been purified after expression in E. coli. In the presence of RNA, CA-NC assembles into regular tube-like structures, with the length of the tube proportional to the length of the RNA (Campbell and Vogt 1995). In these experiments, RNA was required for this type of assembly. These tubes are likely to be similar to the rod-like structures reported for HIV CA itself, but with CA-NC, tube formation occurs at a much lower protein concentration. The NC domain, which is known to contain sequences essential for assembly in vivo (Chapter 7), may promote assembly (both in vivo and in vitro) by binding to RNA and bringing the CA domains and other Gag domains together. In vitro assembly conditions also have been worked out for intact M-PMV Gag protein (Klikova et al. 1995; Sakalian et al. 1996), for HIV Gag protein (Spearman and Ratner 1996), and for ASLV Gag proteins missing the protease domain and including all or parts of the domains upstream of CA (S. Campbell and V.M. Vogt, in prep.). Adding the amino-terminal Gag domains to CA-NC of ASLV changes the morphology of the resulting self-assembled particles from tubular to spherical, with RNA still being required for efficient assembly. It is the p10 domain that somehow triggers this morphological change. Such in vitro reconstitution experiments should provide important information about the functions of CA as well as other virion proteins.
NC Protein
The nucleocapsid (NC) protein is a small basic protein, typically about 60–90 amino acid residues long. Early work in the ASLV and MLV systems showed that this protein is tightly bound to the genomic RNA (Davis and Rueckert 1972; Fleissner and Tress 1973), from which it can be removed by treatment with high concentrations of salt. Treatment of intact virions with UV light crosslinks NC to the genomic RNA (Méric et al. 1984). Because retroviral Gag proteins were initially named by their apparent molecular weight in SDS-polyacrylamide gel electrophoresis or gel filtration in guanidine HCl, because there are several small proteins in virions, and because immunological reagents and sequence information were incomplete, some early reports may have confused NC with the protein derived from sequences between MA and CA (p10 in ASLV and p12 in MLV), or with the transmembrane component of Env (p12E or p15E in MLV), or with MA itself (p15 in MLV and p19 in ASLV). Thus, the older literature on NC should be read with considerable caution. In ASLV, NC was reported to be phosphorylated (Fu et al. 1988), although the first description of phosphoproteins in ASLV (Lai 1976) and the most recent data provide no evidence for phosphorylation of more than a tiny percentage of all the NC molecules in virions (Pepinsky et al. 1996; J.W. Wills, pers. comm.).
In all retroviruses except those of the spumavirus group, NC has one or two characteristic motifs made of regularly spaced cysteine and histidine residues. The retroviral Cys-His motif has the structure CX2CX4 HX4C (here abbreviated CCHC), where most of the residues designated by Xs are not conserved either among retroviruses or between the two motifs of a single NC. An exception is the common placement of an aromatic residue between the first two C residues. The CCHC motif is similar to other short cysteineand histidine-containing structures, called “zinc fingers,” that coordinate a Zn++ ion and that have a role in binding of certain proteins to nucleic acids. Indeed, NC has been shown to bind Zn++ ions tightly both in vitro and in virions (Bess et al. 1992; Chance et al. 1992). Typically, clusters of lysine or arginine residues follow the CCHC motifs. Deletions or major alterations of the CCHC result in the absence of viral RNA in virions or alterations of the specificity of RNA packaging (Chapter 7). Thus, this NC motif probably interacts with the “packaging sequences” near the 5′end of retroviral genomic RNAs when it is still part of the Gag precursor (for review, see Linial and Miller 1990; Berkowitz et al. 1996). In viruses such as HIV-1 and ASLV, which have NC proteins with two CCHC motifs, one of these motifs appears to have the paramount role in RNA encapsidation. NC also contains sequences that act as “assembly domains” (Wills and Craven 1991), which are required for assembly or budding of the virion. The assembly domains are independent of the CCHC and are likely to coincide with the stretches of basic residues.
The absence of CCHC motifs in spumavirus Gag proteins has remained mysterious. However, recent work points to a quite different mode of replication of the prototype virus for this genus, the spumavirus reported to be derived from human cells, human foamy virus (HFV). In this virus, the Pol product is expressed from a spliced RNA and not by frameshifting or termination suppression, reverse transcription appears to take place in virions before infection of a new cell, and Gag may not be cleaved into constituent mature proteins (Konvalinka et al. 1995; Yu et al. 1996). Each of these properties resembles those of hepadnaviruses. It is likely that the lack of CCHC motifs in some way reflects the different biological properties of spumaviruses.
A number of the activities demonstrated for purified NC in vitro have shed light on its function in vivo. NC promotes annealing of complementary RNA sequences (Prats et al. 1988). For example, it stimulates reverse transcription by facilitating the annealing of the primer tRNA to the primer-binding site and promoting strand transfer (Chapter 4). It also stimulates formation of dimeric RNA, presumably by facilitating the pairing of sequences at the dimer linkage. The CCHC motifs appear not to be essential for annealing activity, since deleted versions of NC lacking both of these sequences remain competent in this assay. Rather, it is probably the clustered basic amino acids that provide this function of NC (deRocquigny et al. 1992). Since NC mutations block RNA packaging, it might be expected that NC could specifically recognize the viral encapsidation sequence near the 5′end of the genome. At least in the case of HIV-1, such specific binding has been demonstrated in vitro (Berkowitz et al. 1993; Dannull et al. 1994). Both the runs of basic residues and the CCHC motifs are important for the specific interaction; both motifs also contribute to specific packaging in vivo (Dorfman et al. 1993; Gorelick et al. 1988; Méric et al. 1988). Again, it is important to remember that during the assembly process, NC and all of the other Gag and Pol proteins exist as domains of a larger polyprotein, not as separate polypeptides. It seems likely that the adjoining amino acid sequences may modify the biochemical and functional properties of NC. For example, crosslinking of RNA to the NC domain of Gag in immature virions is much less efficient than crosslinking to the mature NC protein in mature virions (Stewart et al. 1990). In addition, the thermal stabilty of the genomic RNA dimer is lower in the immature virus than in the mature virus (Fu and Rein 1993; Fu et al. 1994), suggesting that the role of NC in promoting RNA-RNA annealing requires that it be proteolytically liberated.
The structures of HIV-1 and MLV NC in solution have been worked out by NMR and other techniques (Fig. 9D) (Morellet et al. 1992; Summers et al. 1992; Demene et al. 1994). In the presence of Zn++, the two CCHC motifs fold into a compact structure. In contrast, the amino-terminal and carboxy-terminal portions of the polypeptide appear to remain flexible. However, it is possible that these portions fold into a more compact structure when NC binds RNA. It is also possible that the sequences near the ends of the NC polypeptide may adopt folded conformations in the context of the uncleaved Gag precursor.
Other Gag Proteins and Peptides
In addition to the proteins discussed above, many retroviral gag genes encode polypeptide segments that lie between MA and CA, between CA and NC, and/or downstream from NC (see Fig. 7). In most cases, the functions of these segments are poorly understood.
The segment of Gag between the ASLV MA and CA regions is processed into three polypeptide fragments. p10 is a proline- and glycine-rich protein. It is preceded by a 22-amino-acid segment often called “p2,” which is processed into two smaller peptides, p2a and p2b (Pepinsky et al. 1986). p2b contains the sequence Pro-Pro-Pro-Tyr, which is found in a large number of retroviral Gag proteins approximately 150 amino acid residues from the amino terminus (see Fig. 7). Mutation or ablation of this motif results in a defect in assembly; apparently, budding is blocked at a late stage (Chapter 7). Processing at the carboxyl terminus of MA is slow, and some viral preparations, especially from some variant strains of RSV, contain the MA-p2 fusion protein, sometimes called p23. The amino-terminal domains of the BLV Gag protein in some ways appear similar to those of ASLV. A longer version of MA, “p15,” is processed slowly into a shorter one (“p10”) plus two peptide fragments (Katoh et al. 1991). MLV encodes a single polypeptide, p12, with an amino acid composition similar to that of ASLV p10, which is processed from the portion of Gag between MA and CA. Early reports that p12 binds specifically to viral RNA have not been confirmed, and it is possible that the RNA-binding protein was actually NC. MLV p12 is phosphorylated, but, as with most phosphorylations of Gag proteins, the function of this modification is unknown. In M-PMV, two proteins, p24 and p12, are derived from the sequence between MA and CA. On the basis of deletion analysis, the latter has been suggested to have a role in assembly of precursor intracytoplasmic A particles characteristic of this virus (Sommerfelt et al. 1992). In MMTV, what might be the same functional sequence as M-PMV p12 is cleaved into several shorter peptides (Hizi et al. 1989).
In at least two genera of retroviruses, the carboxyl terminus of CA and the amino terminus of NC are separated by a short segment sometimes called a “spacer” peptide (SP). In HIV-1, this peptide is 14 amino acids long, and in ASLV, it was originally reported to be 9 amino acids in length. However, more recent data imply that diverse ASLVs contain two species of CA which are present in a constant proportion and which differ by three amino acid residues at their carboxyl termini, implying that the spacer is actually 12 amino acids in length (Pepinsky et al. 1995). The spacer itself may undergo additional cleavage during processing of the Gag protein. During morphogenesis of ASLV and HIV-1, the SP-NC site is the first site to be cleaved, whereas the CA-SP sites are cleaved as a late step (Chapter 7). Deletion of the HIV-1 sequence corresponding to this peptide results in gross aberrations in budding and loss of infectivity (Pettit et al. 1994; Kräusslich et al. 1995). Deletion of this sequence in ASLV leads to the budding of particles that are aberrant in size and are not infectious (Craven et al. 1993; Pepinsky et al. 1995; N.K. Krishna et al., in prep.).
Gag proteins distal to NC are of three types. In a few viruses, such as ASLV, the viral protease forms the carboxy-terminal domain of Gag. Since it is made and incorporated into virions in equimolar amounts with the other structural proteins, it may have a structural role. Mutant particles lacking protease appear to assemble with normal efficiencies and package RNA in some cells (Wills et al. 1991) but not in others (Oertle and Spahr 1990; Stewart and Vogt 1991). In some other viruses, only a few Gag amino acids (four in MLV) follow the carboxyl terminus of NC and are removed during processing.
In HIV-1 and other lentiviruses, a polypeptide of approximately 60 amino acids is cleaved from the Gag protein downstream from NC in a region partially overlapping the pro reading frame. This “p6” domain appears to have a role in release of virus in the final steps of budding and in incorportion of the Vpr and Vpx proteins into the virion (Chapter 7). Viral particles from mutants with p6 deleted or altered remain tethered to the plasma membrane (Göttlinger et al. 1991). The amino acid sequence that functions to allow release of the virus has been mapped to a conserved segment near the amino-terminal domain of p6, and it appears that the late budding defect caused by deletions of this sequence manifests itself only if protease is functional, i.e., not in constructs expressing only Gag (Huang et al. 1995). HIV p6 is considered to be rich in proline residues, but the analogous protein from EIAV is not. Studies of artificially constructed chimeric Gag proteins have shown that when added to the carboxyl terminus of ASLV Gag, HIV p6 can suppress the late budding defects exhibited by mutants of the PPPY motif in p2b (Parent et al. 1995). Like the suppression of ASLV MA deletions by the Src oncoprotein myristylation sequence, this example of the functional exchangeability of a carboxy-terminal and internal sequence attests to the plasticity of the Gag protein, which is all the more remarkable in view of the absence of significant sequence similarities among viruses of different genera.
Proteins Derived from pol and pro
All infectious retroviruses carry three enzymes, reverse transcriptase (RT) and integrase (IN) (Chapter 4) and protease (PR) (Chapter 7) (for review, see Katz and Skalka 1994). The RT protein also contains an additional enzymatic activity, RNase H, which has been mapped to a separate, contiguous portion of the polypeptide, and the conventional designation “RT” always implies the protein with both reverse transcriptase and RNase H activities. The enzymes form domains on the Gag-Pro or Gag-Pro-Pol precursor polypeptide, which was first identified in the avian viruses (Oppermann et al. 1977), although the domains are not always cleaved into separate mature proteins. Examples of how these domains are positioned in prototypic viruses are shown in Figure 8. In most genera, all enzymes are translated together as a Gag-Pro-Pol precursor, which is processed late in assembly to yield the mature forms of the enzymes. Whether expression of pro and pol is by frameshifting or termination suppression, approximately 5% as much RT and IN on a molecular basis is synthesized and packaged into a virion as Gag protein. For most retroviruses, the same holds for PR. As discussed above, in viruses belonging to the type-B, type-D, and HTLV genera, the consequence of a double frameshift is that more PR is synthesized than RT and IN, since it is derived from the Gag-Pro precursor. Since RT, IN, and PR are essential for viral replication and have characteristics that distinguish them from related cellular enzymes, they all have become targets for drug intervention in acquired immunodeficiency syndrome (AIDS) (Chapter 12).

Prior to 1970, enzymes able to make a faithful DNA copy of an RNA strand were unknown. The discovery of RT (Baltimore 1970; Temin and Mizutani 1970) led to a rapid revolution in the understanding of retroviral replication. It also played an important part in the development of the present models for the evolution of life, in which RNA is thought to be both the primordial repository of genetic information and the original enzymatic molecule. Apparently, it was only later in evolution that DNA became used to store information and the more versatile proteins to catalyze chemical reactions (for review, see Gesteland and Atkins 1993). If this view is correct, RTs would have been essential for the transition from an “RNA world” to a “DNA world” (Chapter 8), but whether the viral enzymes are direct descendants of this special molecule remains to be determined.
The first retroviral RTs to be studied were those of ASLV and MLV, which were the major model retroviral systems in the 1970s and early 1980s. Analysis of the biochemistry of RT was facilitated by simple assays, for example, the incorporation of radioactive thymidine triphosphate into DNA in the presence of a poly(A) template and an oligo(dT) primer. The fact that certain cellular polymerases that are not bona fide RTs can also polymerize TTP in the homopolymer assay led to some early incorrect reports of the prevalence of virus-like RT in uninfected cells. Most of this confusion was due to the ability of cellular DNA polymerases to use some RNA templates at low efficiency and was resolved by the use of more discriminating assays, for example, poly(dC) as template with oligo(dG) as primer. A more recently developed and highly sensitive assay, capable of detecting the RT in only a few virions, is based on amplification by polymerase chain reaction of a DNA product of heteropolymeric RNA (Pyra et al. 1994). Although retrovirus-like reverse transcription is not now thought to be a normal feature of eukaryotic cell biology, RTs are known to occupy numerous other biological niches, including the plant caulimoviruses and animal hepadnaviruses, as well as some DNA elements capable of transposition, such as retrotransposons, group II introns, and certain bacterial elements. The only known essential role for reverse transcription in cell growth is the highly specialized repair of chromosomal ends by telomerase.
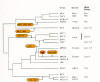
The best studied RTs are from ASLV, HIV-1, and MLV. Like all RTs, they contain highly conserved amino acid sequences diagnostic of this class of enzymes. It is these and other conserved sequences that have been used extensively to construct phylogenetic trees for retroviruses (Doolittle et al. 1989), as shown in Figure 6. Despite the obvious similarity in primary amino acid sequence, the subunit structure of RTs differs among viruses of different genera. In ASLV and HIV-1, RT is a heterodimer in solution, whereas in MLV, RT is a monomer that dimerizes when presented with a template (Chapter 4) (see Fig. 8). The α and β subunits of the ASLV enzyme differ in the cleavage at the RT-IN boundary. The α polypeptide contains only the polymerase and RNase H domains, whereas the β polypeptide contains in addition the IN domain. The HIV-1 dimer structure differs in that the larger (p66) polypeptide is analogous to ASLV RTα, containing both the polymerase and RNase H domains but not IN, and the smaller (p51) subunit is missing the complete RNase H domain. In the HIV heterodimer, biochemical studies show that p51 does not have an active role in catalysis (Le Grice et al. 1991), but it does have a role in the overall structure of the enzyme, consistent with the three-dimensional structure of HIV-1 RT, which shows important differences in folding of the two subunits. At least some retroviral RTs studied bind specifically to the cellular tRNA species used to prime synthesis of the minus strand, and this binding appears to be necessary for the incorporation of tRNAs into the virion (Peters and Hu 1980; Mak et al. 1994)
RNase H was identified initially as an activity of RT that degrades the viral RNA used as a template when permeabilized virions were incubated under conditions where RT could function (Moelling et al. 1971). The “H” refers to the specificity of this enzyme for RNA/ DNA hybrids. Although always part of the RT polypeptide, RNase H forms a distinct subdomain identifiable by both mutational (Tanese and Goff 1988) and structural (Kohlstaedt et al. 1992; Jacobo-Molina et al. 1993) analyses. In vitro, RNase H can function both in the absence of synthesis of DNA by RT and coordinately with it. In the latter case, the RNA portion of the RNA/DNA hybrid formed by RT is destroyed after emerging from catalytic pocket of the RT.
The best studied integrases are those of HIV-1, MLV, and ASLV. This enzyme was first identified as an endonuclease in ASLV virions (Grandgenett et al. 1978), which allowed its purification and characterization. Although subsequent genetic analysis demonstrated the importance of the IN domain for integration of viral DNA (Panganiban and Temin 1984), the role of IN in integration could not be determined until the development of in vitro assays, initially using preintegration complexes containing viral DNA, IN, and some other proteins extracted from newly infected cells (Brown et al. 1987), and later with purified, recombinant IN and simple oligonucleoide substrates (Craigie et al. 1990; Katz et al. 1990). Purified IN has the ability to recognize the ends of the newly synthesized linear double-stranded viral DNA, to remove two nucleotides from the 3′end of each strand, and to join this DNA end to a target DNA at approximately random sites. These activities are consonant with the role IN has in viral replication in vivo (Chapter 5). In comparisons across the retroviral genera, IN is less conserved in amino acid sequence than RT, although certain features, including the D-D-35-E motif of the active site and the three-dimensional structure, seem to be highly conserved.
Many viruses encode proteases, which typically have roles in processing of the primary translation product and maturation of the viral particle (for review, see Dougherty and Semler 1993). The first retroviral PR discovered was that of ASLV (von der Helm 1977). PR acts late in assembly and budding, or perhaps immediately after budding, to sever the domains of Gag and Gag-Pol, causing profound morphological changes and converting the particle into an infectious virus (Chapter 7) (for review, see Vogt 1996). In lentiviruses, PR may have a role not only during virion morphogenesis, but also early in the life cycle upon infection of the cell (Roberts et al. 1991; Nagy et al. 1994). Sequence comparisons of PRs from diverse retroviruses led to the inference that this enzyme is related to cellular aspartic (or “acid”) proteases, which use two apposed aspartate residues in their active site for catalysis. Examples of this widespread class of proteases include the mammalian enzymes pepsin and renin and enzymes from lower eukaryotes (for review, see Davies 1990). Cellular aspartic proteases are monomeric: A single polypeptide composed of two domains of similar amino acid sequence folds into a bilobed structure. In contrast, the three-dimensional structure of ALSV and HIV-1 PRs inferred from crystallography (Lapatto et al. 1989; Navia et al. 1989; Wlodawer et al. 1989; Jaskólski et al. 1990) shows that retroviral proteases are homodimers, with each subunit corresponding to approximately half of the cellular enzyme. As a consequence, dimerization is crucial for enzymatic activity and is likely to be involved in the regulation of proteolysis of Gag and Pol proteins, and therefore essential for proper virion formation. Premature activation of PR in the infected cell leads to premature cleavage of Gag, thus aborting the assembly process (Burstein et al. 1991; Kräusslich 1991). Cellular aspartic proteases are synthesized as zymogens, which are activated by proteolytic removal of an amino-terminal segment of polypeptide. It might therefore be expected that proteolytic processing also plays an important part in activation of retroviral PRs, and there is some evidence in support of such a role.
PRs recognize stretches of amino acids about seven to eight residues in length. Overall hydrophobic sequences are preferred. The specificity of different PRs has been studied both by statistical analyses of known Gag and Pol cleavage sites and cleavage sites in nonviral proteins (Pettit et al. 1991; Poorman et al. 1991) and by experimental testing of large numbers synthetic peptides. Among the generalizations emerging from such studies are that cleavage between tyrosine and proline is common and efficient, whereas cleavage only rarely takes place after isoleucine or valine. The peptide or portion of the protein that is a substrate for PR must be extended in order to fit into the groove in PR in which the active site is located, where it lies as if it were in a β-sheet (see Chapter 7). This relationship between substrate and enzyme suggests that the natural cleavage sites in Gag and Pol are flexible linkers that connect separately folded domains of the polyproteins.
The coding sequence for DU, dut, is found in only two retroviral lineages, the nonprimate lentiviruses and the B-type and D-type viruses (see Fig. 6) (Elder et al. 1992). When present, DU is found in virions and efficiently and specifically degrades deoxyuridine triphosphate, no doubt serving to prevent its incorporation into viral DNA. This enzyme is dispensable for viral replication in dividing cells, but evidence in the EIAV system suggests that it has a role in helping the viruses replicate in quiescent cells such as macrophages (Threadgill et al. 1993). In the lentiviruses, feline immunodeficiency virus (FIV) and EIAV DUs are encoded between the RT and IN domains of Pol, and in M-PMV, DU is encoded upstream of PR, and thus is derived from the Gag-Pro protein> (Fig. 8). The sequences of these two DU proteins are rather different as well, although both are recognizably related to cellular and herpesvirus dUTPases. Given that not all lentiviruses have a DU, these observations suggest that the DU-coding sequence was acquired independently on at least two separate occasions, from the host genome or perhaps from another virus.
Proteins Derived from env
Like all animal viruses that carry a lipid envelope, the surface of retroviral virions is studded with glycoproteins (envelope or Env proteins), whose function is to mediate the adsorption to and the penetration of host cells susceptible to infection (Chapter 3). These proteins were first identified in ASLV and MLV by means of labeling with radioactive sugars (Duesberg et al. 1970) and by the ability of protease digestion to remove them from the surface of virions (Rifkin and Compans 1971). All retroviruses contain two different types of Env proteins, now called SU and TM, that are derived from a common precursor polypeptide (Fig. 10). The proteins often are referred to using the old nomenclature based on apparent molecular weight in SDS-polyacrylamide gel electrophoresis, for example, “gp120” for the SU of HIV. Because of the glycosylation, both the apparent molecular weight in electrophoresis and the actual molecular weight are much greater than that of the polypeptide alone. The genesis of SU and TM and the mechanism of their incorporation into the virion are quite different from those of Gag and Pol. As described above, the messenger for Env is a subgenomic RNA created by splicing, a process that is regulated by viral accessory proteins in the complex viruses. Like cellular proteins destined for secretion, the nascent Env polypeptide binds to a signal recognition particle via its amino-terminal leader segment and then becomes associated with the membrane of the endoplasmic reticulum (ER). There, further translation extrudes most of the polypeptide through the membrane into the lumen of the ER. The protein remains anchored in the membrane by a hydrophobic segment near the carboxyl terminus that spans the membrane once, leaving the carboxy-terminal “tail” of Env in the cytoplasmic compartment. Once in the ER, Env forms the oligomer found in virions, a trimer in the case of ASLV and a multimer of inadequately determined size in HIV. The sequences involved in oligomerization have been mapped in some cases.
After cleavage of the leader sequence, Env is transported by vesicular traffic through the Golgi apparatus to the plasma membrane, in the process becoming N-glycosylated at the consensus sequences Asn-X-Ser or Asn-X-Thr. In some Env proteins, more than two dozen sites for N-glycosylation exist, many of which individually are not essential for biological function (Dedera et al. 1992; Felkner and Roth 1992). The Env proteins of at least some retroviruses also are modified by O-glycosylation (Pinter and Honnen 1988; Bernstein et al. 1994) as well as sulfation (Bernstein and Compans 1992). Env is cleaved while in the Golgi by a cellular protease, either furin or a related enzyme, to yield the mature SU and TM found in virions. Although uncleaved Env proteins are able to bind to the receptor, the cleavage event is necessary to activate the fusion potential of the protein, which is required for entry of the virus into the host cell. SU and TM remain attached to each other by noncovalent interactions, and in some viruses, such as ASLV, also by disulfide bonds. Typically, both SU and TM proteins are glycosylated at multiple sites. However, in some viruses, exemplified by MLV, TM is not glycosylated (Fig. 10).
Once at the plasma membrane, the SU/TM oligomers are incorporated into the budding viral particle (Chapter 7). The cytoplasmic “tail” distal to the membrane-spanning segment of TM remains on the internal side of the viral membrane. This region varies considerably in length in different retroviruses. In some viruses, notably MLV and M-PMV, another cleavage event takes place late in or after budding, resulting in the removal of a short portion of the cytoplasmic tail of TM. This reaction, which is mediated by PR, also has a key role in uncovering the full fusion activity of the protein (Brody et al. 1994; Rein et al. 1994). It has been suggested that the cytoplasmic tail of TM is in contact with the MA domain of the Gag protein in virions, but the nature of this contact is unknown. In other enveloped viruses, for example, alphaviruses (Vaux et al. 1988; Lopez et al. 1994), the cytoplasmic domain of the envelope protein makes a specific and essential contact with an internal virion protein. Analogy suggests that retroviral Env proteins also are likely to be incorporated into virions by this means. However, as discussed above, there is conflicting evidence for MA-TM interaction in retroviruses.
The Env protein is the primary determinant of the type of cell that a retrovirus can infect, because it recognizes the cell surface protein that is the viral receptor (Chapter 3). All enveloped viruses have glycoproteins that bind specifically to receptors on the host-cell membrane. In some cases, these receptors are common and in others, they are rare. An example of the former is sialic acid, found on numerous cell surface proteins, which is recognized by influenza hemagglutinin. Several retroviral receptors have been identified, the best studied of which is the CD4 protein found on helper T lymphocytes and macrophages. CD4 is required for binding and entry of HIV (Dalgleish et al. 1984), although this virus also requires a second, quite different membrane protein for entry. The receptor for ecotropic MLV was cloned using genetically engineered viruses that carry drug resistance markers as a selection for cells that acquired a receptor gene by transfection (Albritton et al. 1989). Similar approaches have led to the identification of receptors for amphotropic MLV, for gibbon ape leukemia virus (GALV), and for subgroup-A ASLV.
Retroviruses are unique among animal viruses in that some groups exhibit considerable polymorphism in receptor usage among otherwise closely related viruses. Relatively minor differences in the ASLV Env sequence can profoundly alter the biological properties of the ASLV protein, changing the receptor to which the virus binds and thus changing its host range. An ASLV is assigned to a particular subgroup—A through E for the most studied viruses—according to the specific receptor used. Susceptibility to infection by a particular subgroup is determined by the single gene in the chicken encoding one of three specific receptors (subgroups B, D, and E recognize allelic receptors). Predicted amino acid sequences of all ASLV Env proteins are quite similar, differing principally in two relatively short segments called host-range-determining (hr) regions. Comparisons of the sequences of env genes encoding different subgroups, combined with studies of recombinant Env proteins, imply that the hr regions combine to interact specifically with the receptor (Dorner and Coffin 1986).
The nomenclature for MLV host range is more complicated, with viruses defined as ecotropic (infecting only mouse cells), xenotropic (infecting only cells other than mouse), polytropic, or amphotropic, depending on the species distribution of suitable receptors. Viruses of both the latter two groups can infect both mouse and non-mouse cells of many species but do so by using different receptors (Rein 1982; Ott and Rein 1992). The sequences that determine the host range have been mapped to the amino-terminal region of SU. Because of the large reservoir of endogenous MLV env genes in the mouse genome (Chapter 8), MLV recombinants carrying altered env sequences arise frequently in the mouse, and these recombinants have a central role in pathogenesis (Chapter 10).
Unlike the ALSVs and MLVs, all strains of HIV and SIV recognize the same receptor, CD4. Nevertheless, major differences in amino acid sequence exist among diverse isolates of these viruses. These differences largely are localized to several variable regions, denoted v1–v4, believed to form loops that extend outward from the central domain of SU. This sequence variation reflects both mutation to escape from the immune system and in vivo variation in biological properties, such as tropism for macrophages or other cell types or ability to form syncytia (Chapter 3). Although the three-dimensional structure of retroviral SU/TM oligomers has not been determined, structural models have been proposed based on several lines of evidence. The location of disulfide bonds, deduced by peptide mapping, suggests that the stretches of polypeptide between the pair of cysteine residues loop out from the core of the molecule. Consistent with this idea, most of the variation in Env sequence occurs in these regions. As expected, sequences involved in receptor interaction have been mapped to the constant regions, principally C2, with contributions from other constant regions as well.
Until a three-dimensional structure is determined, the overall structure and functional organization of retroviral SU and TM proteins can be thought of by analogy with the hemagglutinin (HA) glycoprotein of influenza virus, the best studied of all viral glycoproteins. Although there are some important differences, the mechanism by which HA binds to its receptor and mediates fusion of the viral envelope with the cell's membrane may serve as a paradigm for the function of retroviral Env. Like retroviral Env proteins, HA is a multimer (trimer) cleaved by a Golgi enzyme at a sequence with multiple arginine or lysine residues, to yield a surface protein called HA1 and a transmembrane protein called HA2. Following receptor binding, the HA1-HA2 complex must be activated to promote fusion. In the case of HA, activation causes the complex to undergo a conformational change that impels a previously buried hydrophobic “fusion peptide” near the amino terminus of HA2 outward from the core of the molecule. The fusion peptide is thought to anchor itself into the target membrane and somehow promotes conjoining of the lipid bilayers. For HA, the signal for activation is the low pH of the endosome into which the influenza virion is taken up after receptor binding. For retroviruses, the nature of this signal has not been well established, but at least in most systems, it appears not to involve a pH change (Chapter 3).
Practical considerations make Env proteins more difficult to quantitate than Gag proteins. In almost all retroviruses, much less Env than Gag protein is incorporated into particles. Conventionally, the ratio of Env to Gag is considered to be about 1:10, but in fact this number has not been measured with care for many viruses. Little is known about the factors that control the relative stoichiometry of Env and Gag or the mechanisms by which the Env complex is selectively incorporated into virions. Since it is attached by weak noncovalent interactions, the SU component can be easily lost from virions during their purification. This process of shedding is poorly characterized but has been invoked frequently to explain the low levels of SU in retroviruses. In contrast, the TM component, which is anchored by a transmembrane segment, cannot be removed from virions without their disruption. Finally, virions purified by standard isopycnic centrifugation in sucrose gradients are always contaminated with variable amounts of membrane vesicles sloughed off from live or dead cells. Such membranes, if derived from infected cells, are expected to contain Env protein. Since cells typically synthesize considerably more Env than becomes incorporated into virions, membrane contamination can lead to erroneous estimates of the amount Env in the viral particle.
Other Virus-encoded Proteins in Virions
Products of most retroviral accessory genes (for review of HIV, see Subbramanian and Cohen 1994; Trono 1995; Cohen et al. 1996) are not incorporated into virions. The same is true for the products of oncogenes. The only accessory proteins found in substantial amounts in the viral particle are the related lentiviral products Vpx, present in most SIV strains but not HIV-1, and Vpr, found in all primate lentiviruses. The products of the remaining five HIV and SIV accessory genes are thought to act from within the infected cell, although some evidence suggests that the Vif and Nef proteins may be present in virions as well. However, the numbers of molecules of these proteins are so low—less than about 1% of the Gag molecules—that contamination is difficult to rule out.
The protein products of vpr and vpx are found in large quantities in virions, approaching those of Gag (Cohen et al. 1990). Vpr expressed by itself has a complex distribution in cells, but much of it is in the nucleus. However, when particles are produced at the plasma membrane, Vpr is efficiently recruited into them, and only Gag protein is needed for this recruitment. Deletion mapping points to the carboxy-terminal portion of HIV-1 Gag, namely p6, as the domain responsible for interacting with Vpr (Lu et al. 1993; Paxton et al. 1993; Kondo et al. 1995). The functional parts played by Vpr and Vpx are not understood in detail. Vpr is essential for efficient replication of both HIV-1 and HIV-2 in macrophages and, in addition to MA, is needed for targeting of the newly made viral DNA to the nucleus (Heinziger et al. 1994). This targeting function appears to be critical for the establishment of HIV infection in some nondividing cells, which is a characteristic feature of lentiviruses. Vpr also seems to have a role later in infection, in affecting transit of the cell through the cell cycle.
Although other accessory proteins are not incorporated into virions in substantial amounts, three HIV-1 proteins besides Vpr appear to affect the structure, morphogenesis, or biological function of the mature viral particle and therefore deserve mention here. The vif gene is needed for efficient replication of the virus in primary CD4 cells and in some but not all established cell lines. Vif affects the infectivity of released viral particles; i.e., the requirement for this protein depends on the cells from which the virus is released, rather than on the cells being infected (Gabuzda et al. 1992; von Schwedler et al. 1993). The cell lines that support replication of vif-defective HIV-1 thus appear to be able to supply a “Vif-like” function. In the restrictive cells, it is not the level of virion formation that is reduced in the absence of Vif, but rather the specific infectivity of the particles. This phenotype suggests that the virions are modified in some way by Vif. It has been reported that vif - and vif + virions show ultrastructural differences in the core (Höglund et al. 1994). The mechanism of action of Vif remains unknown.
The vpu gene is found in HIV-1 and very closely related viruses but not in other primate or nonprimate lentiviruses. The product of this gene is a small integral membrane protein. Vpu downregulates the levels of the CD4 receptor by accelerating its destruction. This activity is carried out in association with the endoplasmic reticulum or other membranes internal to the cell, and it requires association between SU and CD4 (Willey et al. 1992). However, Vpu also promotes release of the budding virion at the plasma membrane (Göttlinger et al. 1993) (Chapter 7). Vpu action is not specific for HIV-1, since it enhances release of other lentiviruses as well as MLV. The mechanisms underlying this final stage in budding are not known for any enveloped virus.
The product of the HIV nef gene is also membrane-associated. Nef has complex effects on signal transduction pathways in the cell and, like Vpu, leads to loss of the CD4 receptor, in this case directly from the cell surface (Garcia et al. 1993). An additional result of Nef expression is the increased specific infectivity of viral particles (Schwartz et al. 1995). Virions released from cells in the presence or absence of Nef are indistinguishable in number, in the amounts of Gag and Env protein, and in the amount of reverse transcriptase activity measured by standard exogenous assays. However, virions of nef + virus are capable of more viral DNA synthesis, suggesting that Nef directly or indirectly activates reverse transcriptase. The low levels of Nef that appear to be associated with virions could be responsible for this phenomenon. That Nef is a bona fide virion constituent is supported by the observation that some of this protein is found to have been cleaved by PR (Welker et al. 1996).
Cellular Proteins in Virions
Even the purest samples of retroviral particles show a plethora of cellular polypeptides by SDS-polyacrylamide gel electrophoresis when a large enough quantity of virus is analyzed. Thus, it is not surprising that numerous enzymatic activities have been reported in viral preparations. One of the earliest enzymes associated with retroviral particles was found in AMV (de Thé et al. 1964), a virus usually obtained from infected chicks, where it is shed into the plasma in large amounts by the transformed myeloblasts. Highly purified AMV has a very active ATPase that can be assayed without disruption of the virion, and which has been used as a marker for viral purification. The absence of ATPase from AMV grown in fibroblasts implies that the enzyme is not a virus-coded protein, but rather a component of the myeloblast membrane incorporated into virions during budding.
The biological significance of such host polypeptides is difficult to ascertain, since virions are contaminated with membranous debris from cells, and since a small volume of the cytoplasm may be incorporated passively during budding. Proteins present in small amounts are not necessarily irrelevant, however. Several criteria have been used to gauge if a host protein in a virus may be important for the viral life cycle. First, is the protein enriched in the virus, relative to the cell? Second, is the protein found in some retroviral species but not others grown in the same cells? Third, in a collection of closely related cellular proteins, are only some found in the virus? A positive answer in any of these cases suggests specific interactions with viral proteins during assembly, and specific interactions among proteins in biology usually imply biological function. Fourth, in cases where the level of the protein in the virus can be manipulated, does this affect infectivity? A positive answer again suggests the protein plays a part in infection or replication.
According to these criteria, only a few host proteins have been implicated as important virion constituents, including both cytoplasmic and membrane proteins. In the former category, perhaps the most interesting is cyclophilin A. The cyclophilins are highly conserved proteins that have prolyl isomerase activity and that serve as chaperonins to aid correct protein folding. The naming of these proteins derives from their initial identification as targets for the immunosuppressive drug cyclosporin. Cyclophilins A and B were identified following a systematic search for human proteins capable of tightly binding to HIV-1 Gag protein (Luban et al. 1993). The molecular genetic approach used, based on the yeast “two-hybrid” system, allows one to screen a cDNA library for genes that encode proteins capable of binding tightly to a known protein. This now widely used method has also been used to identify host proteins that interact with integrase (Kalpana et al. 1994) and also to study the interaction of Gag proteins with each other (Franke et al. 1994b). HIV-1 Gag also binds to cyclophilins A and B in vitro, confirming the initial result in yeast. Deletion analysis implies that the CA domain of Gag is responsible for this binding. Wild-type HIV-1 virions contain substantial amounts of cyclophilin A (Franke et al. 1994a; Thali et al. 1994). A role for cyclophilin in HIV replication is more directly implied by the observation that cyclosporin treatment of HIV-1-producing cells leads to a large reduction in titer of infectious particles but not of total particles. Virions resulting from this treatment are largely lacking in cyclophilin A. As in the case of Vif, the biological function of cyclophilins is not known. Gag-cyclophilin interaction is specific for HIV-1. HIV-2 does not contain this protein, and its replication is not affected by cyclosporin treatment.
Another host protein that appears to be a real constituent of virions is ubiquitin, a small highly conserved protein found in all eukaryotes. This protein has multiple functions in the cell. For one, its covalent attachment to lysine residues of a host protein marks that protein for degradation by the proteosome pathway (for review, see Ciechanover 1994). Ubiquitin also becomes covalently conjugated to histones and to certain cell surface receptors. Free ubiquitin has been found in ASLV, at a concentration greater than that in the bulk cytoplasm of cells, or about 100 molecules per virion (Putterman et al. 1990). This protein also has been found in HIV-1 and MLV, both free and conjugated to Gag proteins (L.A. Henderson, pers. comm.). A number of different viruses interact with ubiquitin pathways in their life cycle. Baculoviruses carry a gene for a ubiquitin variant that is attached to the virion membrane by a phospholipid anchor (Guarino et al. 1995). Some strains of bovine infectious diarrhea virus, a pestivirus, carry a partial ubiquitin gene derived from the host and fused to a gene encoding a virion protein. This fusion protein is somehow responsible for greatly increased virulence (Meyers et al. 1991). The possible functional role of ubiquitin in the retroviral life cycle has not been explored.
Of the other proteins that have been noted in preparations of retroviral particles, perhaps the cytoskeletal proteins are the most suggestive of function. For example, actin has been commonly seen in small amounts in preparations of retroviruses and other enveloped viruses, but because of its abundance in the cell, it is frequently dismissed as an artifact. However, in the case of HIV, biochemical evidence that Gag binds to F-actin has been presented (Rey et al. 1996). In addition, actin and other cytoskeletal proteins have been documented as genuine virion constituents, as indicated by the presence of several proteolytic cleavage fragments generated by the action of PR (Ott et al. 1996). It is tempting to speculate that filamentous actin could play a part in the budding process, as was suggested for RSV and MMTV many years ago (Damsky et al. 1977) (Chapter 7). However, these possibilities have not been examined critically.
In view of the phenomenon of pseudotyping, in which retroviruses incorporate the envelope protein of another virus (Chapter 3), it is not surprising that some host membrane proteins also are picked up during the budding process. Quantitative studies of host membrane proteins in virions have been reported only infrequently. If the human CD4 protein is expressed in quail cells, it can be efficiently incorporated into ASLV virions budding from the same cells (Young et al. 1990). Although the biological significance of this artificial system is uncertain, it does indicate that nonspecific incorporation of any surface protein is possible, perhaps requiring only that the protein be unattached to internal cell components (Chapter 3).
Incorporation of cellular proteins into virions can be unimportant for viral replication yet still have important practical consequences. For example, HIV-1 includes large amounts of the major histocompatibility complex (MHC) class I proteins on its surface during budding. These surface glycoproteins, which have key roles in immune recognition, are present in virions in quantities not very different from those of the viral Env protein (Arthur et al. 1992). Although contaminating membrane vesicles would be expected to carry some MHC proteins, the observation that some MHC forms but not others copurify with HIV-1 suggests specificity. The presence of these proteins on virions is demonstrated by immunoprecipitation of Gag protein by treatment of intact virions with anti-class I antibody. That these proteins can be biologically important is shown by the observation that immunization by human class I protein protects monkeys from infection by SIV grown on human cells and does so much more efficiently than immunization with SIV virions grown on monkey cells (Arthur et al. 1995) (Chapter 12). These findings could have further importance for the understanding of HIV pathogenesis (Chapter 11). The principles by which membrane components such as MHC proteins are selected by the virus during the budding process remain mysterious.
- Virion Proteins - RetrovirusesVirion Proteins - Retroviruses
Your browsing activity is empty.
Activity recording is turned off.
See more...