Summary
Clinical characteristics.
Xeroderma pigmentosum (XP) is characterized by:
- Acute sun sensitivity (severe sunburn with blistering, persistent erythema on minimal sun exposure) with marked freckle-like pigmentation of the face before age two years;
- Sunlight-induced ocular involvement (photophobia, severe keratitis, atrophy of the skin of the lids, ocular surface neoplasms);
- Greatly increased risk of sunlight-induced cutaneous neoplasms (basal cell carcinoma, squamous cell carcinoma, melanoma) within the first decade of life.
Approximately 25% of affected individuals have neurologic manifestations (acquired microcephaly, diminished or absent deep tendon stretch reflexes, progressive sensorineural hearing loss, progressive cognitive impairment, and ataxia). The most common causes of death are skin cancer, neurologic degeneration, and internal cancer. The median age at death in persons with XP with neurodegeneration (29 years) was found to be younger than that in persons with XP without neurodegeneration (37 years).
Diagnosis/testing.
The diagnosis of XP is established in a proband on the basis of clinical findings and family history and/or by the identification of biallelic pathogenic variants in DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, or XPC.
Management.
Treatment of manifestations: Premalignant skin lesions such as actinic keratoses can be treated by freezing with liquid nitrogen; larger areas can be treated with field treatments such as topical 5-fluorouracil or imiquimod preparations. Rarely, therapeutic dermatome shaving or dermabrasion has been used; skin neoplasms can be treated (as in persons without XP) with electrodesiccation and curettage or surgical excision; skin cancers that are recurrent or in locations at high risk for recurrence are best treated with Mohs micrographic surgery. Oral isotretinoin or acitretin can prevent new skin neoplasms but have many side effects. Neoplasms of the eyelids, conjunctiva, and cornea can be treated surgically; corneal injury associated with eyelid abnormality can be decreased with eye drops or soft contact lenses; corneal transplantation may improve the visual impairment resulting from severe keratitis. Hearing loss may be treated with hearing aids.
Prevention of primary manifestations: Avoid sun and other UV exposure to the skin and eyes. Measure UV light with a light meter in an affected individual's home, school, and/or work environment so that high levels of environmental UV can be identified and eliminated.
Surveillance: Skin examinations by a physician every three to 12 months; eye exams for signs of UV exposure and damage every six months; routine eye and neurologic examinations for progressive neurologic abnormalities every 12 months; audiograms every six to12 months.
Agents/circumstances to avoid: UV exposure from sunlight and artificial sources of UV radiation; cigarette smoke.
Evaluation of relatives at risk: If family-specific pathogenic variants have been identified, molecular genetic testing of at-risk sibs can permit early diagnosis and rigorous sun protection from an early age.
Pregnancy management: Systemic retinoids (isotretinoin, acitretin) may be used as skin cancer chemopreventive agents. These drugs are known to be teratogenic to a developing fetus and pose a high risk for birth defects. Women of reproductive age who are taking a systemic retinoid must use effective contraception and be monitored with regular pregnancy tests.
Genetic counseling.
XP is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an XP-related pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the XP-related pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for XP are possible.
Diagnosis
Moriwaki et al [2017] (full text) published diagnostic criteria focused on Japanese individuals with xeroderma pigmentosum (XP).
Suggestive Findings
XP should be suspected in individuals with the following clinical findings and family history.
Clinical findings
- Skin
- Acute sun sensitivity (severe sunburn with blistering or persistent erythema on minimal sun exposure)
- Marked freckle-like pigmentation (lentigos) on the face before age two years
- Skin cancer within the first decade of life
- Eye
- Photophobia with prominent conjunctival injection
- Severe keratitis, sometimes resulting in corneal opacification and vascularization
- Increased pigmentation of the lids with loss of lashes
- Atrophy of the skin of the lids resulting in ectropion, entropion, or in severe cases complete loss of the lids
- Ocular surface neoplasms
- Nervous system
- Diminished or absent deep tendon stretch reflexes. EMG and nerve conduction velocities may show an axonal (or mixed) neuropathy.
- Progressive sensorineural hearing loss. Audiometry may reveal early high-tone hearing loss.
- Acquired microcephaly. CT and MRI of the brain may show enlarged ventricles with thinning of the cortex and thickening of the bones of the skull.
- Progressive cognitive impairment
- Ataxia
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of XP is established in a proband on the basis of clinical findings and family history (see Suggestive Findings) and/or by the identification of biallelic pathogenic variants in one of the genes listed in Table 1.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of XP has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
A xeroderma pigmentosum multigene panel that includes all of the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
NOTE: Some regions of the world have increased prevalence of XP and the proportion of XP attributed to pathogenic variants in a specific gene may vary by country of origin of the proband associated with particular founder variants in that region. See Prevalence.
Table 1.
Molecular Genetic Testing Used in Xeroderma Pigmentosum
Clinical Characteristics
Clinical Description
The findings from 106 individuals with XP examined at the National Institutes of Health (US) in a long-term study from 1971 to 2009 by Bradford et al [2011] and Fassihi et al [2016] are summarized in Table 2 and the text following.
Table 2.
Xeroderma Pigmentosum: Frequency of Select Features
Cutaneous findings. More than half of individuals with XP have a history of acute sunburn reaction on minimal UV exposure. The remainder of individuals with XP tan without excessive burning [Sethi et al 2013]. In all individuals, numerous freckle-like hyperpigmented macules appear on sun-exposed skin.
- The median onset of the cutaneous symptoms is between ages one and two years.
- These abnormalities are limited to sun-exposed areas.
- Continued sun exposure causes the skin to become dry and parchment-like with increased pigmentation; hence the name xeroderma pigmentosum ("dry pigmented skin").
- Most individuals with XP develop xerosis (dry skin) and poikiloderma (the constellation of hyper- and hypopigmentation, atrophy, and telangiectasia).
- Premalignant actinic keratoses develop at an early age.
- XP is an example of accelerated photoaging. The appearance of sun-exposed skin in children with XP is similar to that occurring in farmers and sailors after many years of extreme sun exposure [Rizza et al 2021].
Ocular abnormalities are almost as common as the cutaneous abnormalities [Brooks et al 2013]. Beginning in the first decade of life, findings are usually limited to the anterior, UV-exposed portions of the eyes including the conjunctiva, cornea, and lids.
- Dry eyes are a common finding in XP and may be seen in patients in the first decade of life.
- Epithelioma, squamous cell carcinoma, and melanoma of UV-exposed portions of the eye are common.
- The ocular manifestations may be more severe in heavily pigmented individuals.
- Benign conjunctival inflammatory masses that develop can spread to obscure the cornea [Mahindra et al 2008].
- Corneal findings include photophobia, severe keratitis, corneal opacification, and neovascularization.
- Lid findings include atrophy of the skin of the lids resulting in ectropion, entropion, or (in severe cases) complete loss of the lids. Lentigines, freckling on the lids, and lash loss are also common findings.
Neurologic findings. Progressive neurologic abnormalities that worsen slowly were reported in approximately 25% of 106 affected individuals.
- The onset may be early in infancy or, in some individuals, delayed until the second decade or later [Rapin et al 2000, Shanbhag et al 2018].
- The neurologic abnormalities may be mild (e.g., isolated hyporeflexia) or severe, including acquired microcephaly, progressive intellectual impairment, sensorineural hearing loss beginning with high frequencies, spasticity, ataxia, and/or seizures.
- During an upper respiratory infection some individuals may develop difficulty swallowing or, rarely, vocal cord paralysis [Ohto et al 2004].
- Reduced nerve conduction velocity may also be present on nerve conduction studies [Lehky et al 2021].
Cutaneous neoplasia. If aggressive UV avoidance is not begun early, accumulated sunlight-induced DNA damage is likely to result in skin cancer within the first decade of life. Bradford et al [2011] found that individuals with XP younger than age 20 years were at increased risk for the following cancers:
- Non-melanoma (basal cell and squamous cell) skin cancer at UV-exposed sites. The >10,000-fold increased risk was associated with a median age of onset of nine years, nearly 60 years earlier than in the US general population.
- Cutaneous melanoma. The >2,000-fold increased risk was associated with a median age of onset of 22 years, more than 30 years earlier than in the US general population.Surprisingly, those with XP who had the most severe sun sensitivity had a later onset of skin cancer – perhaps because they used greater sun protection.
Other neoplasias
- A substantial number of people with XP have oral cavity neoplasms, particularly squamous cell carcinoma of the tip of the tongue, a presumed sun-exposed location [Kraemer et al 1994, Butt et al 2010].
- Gliomas of the brain and spinal cord, tumors of the lung, uterus, breast, pancreas, stomach, kidney, and testicles, and leukemia have been reported in a few individuals with XP [DiGiovanna et al 1998, Bradford et al 2011, Lai et al 2013, Fassihi et al 2016, Sarasin et al 2019b, Oetjen et al 2020].
- Because some of the carcinogens in cigarette smoke bind to DNA, resulting in damage that is repaired by the nucleotide excision repair system, this unrepaired DNA damage may contribute to the development of lung cancer in individuals with XP who smoke. The risk for lung cancer due to exposure from secondhand smoke has not been determined.
Overall, there is an estimated 34-fold increase in internal neoplasms in XP, and tumors arise 50 years earlier compared to the US general population [Nikolaev et al 2022].
Other medical concerns
- Women with XP are at increased risk for premature menopause (menopause before age 40 years) and may require assisted reproductive technology to experience pregnancy [Authors, personal communication]. A study of reproductive health in women with XP identified premature menopause in 31% of the participants, the majority of whom had pathogenic variants in XPC [Merideth et al 2019].
- Individuals with XP are at risk for thyroid nodules and carcinoma. Kouatcheu et al [2021] reported on 29 individuals seen prospectively as part of a natural history study and found that 18 had thyroid nodules and two were diagnosed with papillary thyroid cancer. In addition, researchers studying XP in northern Africa have noted thyroid nodules and thyroid cancer in multiple individuals with XP [Ben Rekaya et al 2013, Hadj-Rabia et al 2013, Jerbi et al 2016]. A 74-fold increased frequency of thyroid cancers was estimated by Nikolaev et al [2022].
Life span. The median age at death (29 years) in persons with XP with neurodegeneration was younger than that in persons with XP without neurodegeneration (37 years) (p=0.02). The three most common causes of death were skin cancer, neurologic degeneration, and internal cancer [Bradford et al 2011]. Early diagnosis and use of sun protection have been shown to extend the life span in Japanese patients [Nakano et al 2016].
Phenotype Correlations by Gene
Table 3.
Xeroderma Pigmentosum: Phenotype Correlations by Gene
Genotype-Phenotype Correlations
No genotype-phenotype correlations, besides those shown in Table 3, have been identified.
Nomenclature
Xeroderma pigmentosum was first described in Vienna by Moriz Kaposi in the textbook of dermatology he published in 1870 with his father-in-law, Ferdinand Hebra. The disorder was first called xeroderma or parchment skin. See discussion in Kraemer et al [1987] and in DiGiovanna & Kraemer [2012].
Previously, an individual with XP with any neurologic abnormality was said to have DeSanctis-Cacchione syndrome. With clarification of the spectrum of XP disease, this term is now reserved for XP with severe neurologic disease, dwarfism, and immature sexual development. The complete DeSanctis-Cacchione syndrome has been recognized in very few individuals; however, many individuals with XP have one or more of its neurologic features.
"Pigmented xerodermoid" is now known to be identical to the XP variant.
Before the genes responsible for XP were identified, complementation groups were used to categorize functional defects in affected individuals. In an XP complementation analysis, cells from affected individuals were fused in the laboratory to determine whether their defects were different, in which case they would be able to supply all functions necessary to restore a normal cellular phenotype. Complementation is therefore a test of function and enabled the categorization of affected individuals as having the same or different defects. Subsequently, each complementation group was found to result from a defect in a distinct gene [DiGiovanna & Kraemer 2012]. Testing to assign complementation group is not available on a clinical basis, but the complementation group names are used clinically to describe the different phenotypes associated with underlying genes (see Table 1).
Prevalence
Prevalence is estimated at 1:1,000,000 in the United States and Europe [Kleijer et al 2008].
Certain populations have a higher prevalence. This is usually related to the presence of founder variants (see Table 9):
- In Japan prevalence is estimated at 1:22,000 [Hirai et al 2006].
- In North Africa (Tunisia, Algeria, Morocco, Libya, and Egypt) [Ben Rekaya et al 2009, Messaoud et al 2010, Soufir et al 2010] and the Middle East (Turkey, Israel, and Syria) [Kraemer & Slor 1985, Jerbi et al 2016] prevalence is increased, as high as 1:10,000, especially in communities in which consanguinity and endogamy are common [Sarasin et al 2019a].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in DDB2, POLH, XPA, or XPC.
In addition to the xeroderma pigmentosum (XP) phenotypes discussed in this GeneReview, pathogenic variants in ERCC1, ERCC2, ERCC3, ERCC4, and ERCC5 are known to be associated with cerebrooculofacioskeletal (COFS) syndrome, Fanconi anemia, and trichothiodystrophy (TTD) (Figure 1, Table 4). (Note: COFS syndrome and TTD should be considered in the differential diagnosis of XP; see Table 5.)
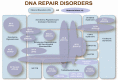
Figure 1.
Relationship between genotype and phenotype in the xeroderma pigmentosum-Cockayne syndrome-trichothiodystrophy spectrum Modified from DiGiovanna & Kraemer [2012]
Table 4.
Allelic Disorders
Complex phenotypes
- Allelic complex phenotypes in the differential diagnosis of XP are summarized in Table 5.
- One individual with phenotypic features of Cockayne syndrome, XP, and Fanconi anemia had biallelic pathogenic variants in ERCC4 [Kashiyama et al 2013].
- Niedernhofer et al [2006] described a male age 15 years with biallelic ERCC4 pathogenic variants and cachexia, dwarfism, microcephaly, marked sun sensitivity from birth, visual impairment (due to optic atrophy), hearing loss, mild learning disabilities, progressive growth failure, facial features characteristic of premature aging, mild ataxia, and poor coordination. Niedernhofer et al [2006] proposed that this represented a new progeroid syndrome (see OMIM 610965). Whether XFE progeroid syndrome represents a distinct condition or is part of the spectrum of XP, Fanconi anemia, or XP / Cockayne syndrome has not been determined.
Sporadic tumors. Investigation of the association between an increased cancer risk and heterozygosity for an allelic variant causing XP is an active area of research. In addition, sporadic tumors occurring as single tumors in the absence of any other findings of XP may harbor somatic variants in XP-related genes.
Differential Diagnosis
Other nucleotide excision repair disorders associated with cutaneous photosensitivity to consider in the differential diagnosis of xeroderma pigmentosum (XP) are summarized in Table 5 (see also Figure 1) [Horibata et al 2004, Berneburg & Kraemer 2007, Kraemer et al 2007, Stefanini & Kraemer 2008, Kraemer & Ruenger 2012, Ruenger et al 2012].
Table 5.
Autosomal Recessive Nucleotide Excision Repair Disorders Exhibiting Cutaneous Photosensitivity
Other. In addition to disorders sharing deficient nucleotide excision repair, other conditions exhibiting cutaneous photosensitivity may be considered in the differential diagnosis of XP, especially in individuals with a paucity of other clinical findings. These include the following:
- Rothmund-Thomson syndrome (RTS). RTS is characterized by a rash that progresses to poikiloderma; sparse hair, eyelashes, and/or eyebrows; small size; skeletal and dental abnormalities; juvenile cataracts; and an increased risk for cancer, especially osteosarcoma. A variety of benign and malignant hematologic abnormalities have been reported in affected individuals. The rash of RTS typically develops between ages three and six months (occasionally as late as age 2 years) as erythema, swelling, and blistering on the face, subsequently spreading to the buttocks and extremities. RTS is caused by pathogenic variants in ANAPC1 or RECQL4 and is inherited in an autosomal recessive manner.
- Hartnup disorder (OMIM 234500). Affected individuals may have reduced levels of niacin with resulting pellagra-like symptoms of photosensitivity with dermatitis, diarrhea, and dementia. However, individuals with Hartnup disorder are not reported to have increased frequency of skin cancer, as is seen in those with XP. Hartnup disorder is caused by pathogenic variants in SLC6A19 and is inherited in an autosomal recessive manner.
- The cutaneous findings of Carney complex may be confused with those of XP; however, Carney complex is characterized by lentigines without evidence of the usually associated signs of sunlight-induced skin damage such as atrophy and telangiectasia (i.e., poikiloderma), and cutaneous findings are not limited to sun-exposed sites [Correa et al 2015].
Management
Evaluations Following Initial Diagnosis
General clinical care guidelines for individuals with xeroderma pigmentosum (XP) have been proposed by the Japanese Dermatological Association [Moriwaki et al 2017] (full text).
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with XP, the evaluations summarized in Table 6 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 6.
Recommended Evaluations Following Initial Diagnosis in Individuals with Xeroderma Pigmentosum
Treatment of Manifestations
Table 7.
Treatment of Manifestations in Individuals with Xeroderma Pigmentosum
Prevention of Primary Manifestations
Treatment of XP depends on early diagnosis and immediate, aggressive avoidance of sun and other UV exposure. This involves avoiding or minimizing outdoor exposure at times when UV radiation is present (when the sun is out or during daytime through clouds).
- Clinical suspicion of XP should prompt immediate sun-protective measures until the diagnosis is confirmed or an alternative explanation is determined.
- Individuals should be educated to protect all body surfaces from UV radiation by wearing protective clothing including hats, long sleeves, long pants and gloves, broad-spectrum, high-SPF sunscreens, UV-absorbing glasses, and long hair styles. Multiple layers of clothing are preferred. The eyes should be protected by wearing UV-absorbing glasses with side shields. Some individuals have custom-made hats with UV-absorbing face shields to permit visibility outdoors while protecting the face from UV.
Because the cells of individuals with XP are hypersensitive to UVA and UVB (found in sunlight) and UVC (found in some artificial light sources), it is useful to measure UV light in an individual's home, school, or work environment with a light meter so that high levels of environmental UV (e.g., halogen lamps) can be identified and eliminated if possible. While no standards exist for perfectly safe UV exposure in individuals with XP, the use of UV meters can alert individuals to unexpected sources of high levels of environmental UV. Unlike UVB, UVA is not blocked by window glass. Windows in areas where individuals with XP will be spending large amounts of time should have UV blocking film applied.
Low vitamin D levels can result from aggressive avoidance of sun exposure. Vitamin D is produced in the skin by a reaction involving exposure to UV radiation. Active adults with XP and skin cancers received sufficient vitamin D in their diet in the past to result in normal serum concentrations of the active form (1,25 dihydroxy vitamin D) [Sollitto et al 1997]. However, children protected from sunlight very early in life have had low serum concentration of 25 hydroxy vitamin D; one child became susceptible to bone fractures [Ali et al 2009; Author, personal observation]. Serum vitamin D levels should be monitored and dietary supplementation with oral vitamin D is recommended for persons with low serum concentration of serum vitamin D [Reichrath 2007; Author, personal communication].
Surveillance
Table 8.
Recommended Surveillance for Individuals with Xeroderma Pigmentosum
Agents/Circumstances to Avoid
UV exposure from sunlight and artificial sources of UV radiation should be avoided (see Prevention of Primary Manifestations).
Artificial sources of UV. Certain light sources (e.g., mercury arc, halogen, and other lamps) can be unrecognized sources of UV. Although such light sources are often shielded, in open areas such as gymnasiums they can be a source of UV if the shield has been breached. UV meters are readily available to enable monitoring of areas to identify unexpected UV sources.
Cigarette smoke. Because cells from individuals with XP are also hypersensitive to environmental mutagens, such as benzo[a]pyrene found in cigarette smoke, prudence dictates that individuals with XP should be protected against these agents. One individual with XP who smoked cigarettes for more than ten years died of bronchogenic carcinoma of the lungs at age 35 years [Kraemer et al 1994]. The authors have cared for another individual with XP who smoked and developed lung cancer in the fifth decade of life.
Evaluation of Relatives at Risk
It is appropriate to evaluate the apparently asymptomatic older and younger sibs of an affected individual by molecular genetic testing in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures. Treatment of XP depends on early diagnosis and immediate, aggressive avoidance of sun and other UV exposure (see Prevention of Primary Manifestations).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
The systemic retinoids isotretinoin and acitretin are used as skin cancer chemopreventive agents in individuals who are actively developing large numbers of skin cancers, and thus may be used by some women with XP [Kraemer et al 1988]. Systemic retinoids are known to be teratogenic to a developing fetus and pose a high risk for birth defects. Therefore, women who are using systemic retinoids should be appropriately counseled about pregnancy risks and the need for effective contraception; regular monitoring with pregnancy tests is indicated. Systemic retinoids should be administered only by physicians who are knowledgeable regarding their risks and benefits.
To access isotretinoin in the US, women and their prescribing providers must be enrolled in the iPLEDGE program to minimize the potential for fetal exposure. Pregnancy avoidance is initiated before therapy, continues during therapy, and extends post-treatment until the drug is cleared from the body. While both isotretinoin and acitretin may be effective in preventing skin cancers, acitretin may take longer to be eliminated from the body, requiring an extended period (3 years) of post-therapy pregnancy avoidance to minimize teratogenic risk.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Xeroderma pigmentosum (XP) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an individual with XP are presumed to be heterozygous for an XP-related pathogenic variant.
- If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an XP-related pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an XP-related pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
- Unless an individual with XP has children with an affected individual or a carrier, his/her offspring will be obligate heterozygotes (carriers) for an XP-related pathogenic variant.
- The offspring of an individual with XP and an individual who is heterozygous for an XP-related pathogenic variant in the same gene as the proband have a 50% chance of having XP. This is a consideration in populations with a founder variant or with a high rate of consanguinity (see Prevalence and Table 9).
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an XP-related pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the XP-related pathogenic variants in the family (see Christen-Zaech et al [2009] for discussion).
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Once the XP-related pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for XP are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Action for XPWestfield, Cushnie, Nr Alford, Aberdeenshire, AB33 8LPUnited KingdomEmail: support@actionforxp.org
- Enfants de la LuneSiège de l’association3 Rue Corneille 01200 Bellegarde sur ValserineFrancePhone: 04 57 05 13 61Email: contact@enfantsdelalune.org
- Xeroderma Pigmentosum Society, Inc (XP Society)437 Syndertown RoadCraryville NY 12521Phone: 877-XPS-CURE (877-977-2873); 518-851-2612Email: xps@xps.org
- XP Family Support Group10259 Atlantis DriveElk Grove CA 95624Phone: 916-628-3814Email: contact@xpfamilysupport.org
- XP Freunde die MondscheinkinderPostfach 212448550 SteinfurtGermanyPhone: 49 176 200 109 30Email: info@xerodermapigmentosum.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Xeroderma Pigmentosum: Genes and Databases
Table B.
OMIM Entries for Xeroderma Pigmentosum (View All in OMIM)
Molecular Pathogenesis
An intact DNA repair system that senses, excises, and repairs UV-induced dipyrimidine photoproducts and other forms of DNA damage is necessary to prevent replication errors and subsequent tumorigenesis (Figure 2) [DiGiovanna & Kraemer 2012].
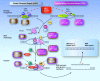
Figure 2.
Nucleotide excision repair (NER) pathway Modified from DiGiovanna & Kraemer [2012]
Exposure to UV radiation from sunlight forms cyclobutane dimers or other photoproducts at adjacent pyrimidines, thereby distorting the DNA. Initial lesion recognition in non-transcribed DNA (global genome repair-GGR) is performed by DDB2-encoded protein [Clement et al 2010, Sugasawa 2010]. The XPC-encoded protein binding to the photoproducts is facilitated by the binding of the DDB2-encoded protein. The XPC-encoded protein is complexed with hHR23B and centrin [Sugasawa 2010].
DNA damage in transcribed genes (transcription coupled repair) is marked by stalled RNA polymerase. The CS (Cockayne syndrome)-encoded proteins (along with others) bind to the damage in the transcribed DNA strand.
ERCC2 is part of basal transcription factor TFIIH that is involved in regulation of the basal rate of transcription (RNA synthesis) of active genes, as well as in nucleotide excision repair (NER).
In both global genome repair and transcription-coupled repair, the XPA protein probably functions in conjunction with replication protein A and TFIIH – the basal transcription factor that is involved in regulation of the basal rate of transcription (RNA synthesis) of active genes, as well as in NER. The XPB/ERCC3 and XPD/ERCC2 proteins (helicases that are part of the TFIIH complex) partially unwind the DNA in the region of the damage, thereby exposing the lesion for further processing. The XPF/ERCC4 product, in a complex with ERCC1, makes a single-strand nick at the 5' side of the lesion, while the XPG/ERCC5 product makes a similar nick on the 3' side, resulting in the release of a region of approximately 30 nucleotides containing the damage. The resulting gap is filled by DNA polymerase using the other (undamaged) strand as a template in a process involving proliferating cell nuclear antigen. DNA ligase I seals the region, restoring the original undamaged sequence [van Steeg & Kraemer 1999, Bootsma et al 2002].
Individuals with the XP variant have a normal nucleotide excision pathway and a defect in POLH, encoding the DNA polymerase eta protein, which can replicate through UV-damaged DNA. Loss of polymerase eta function leads to replication by alternative error-prone polymerases.
Mechanism of disease causation. XP occurs via a loss-of-function mechanism of any of the NER or polymerase eta proteins.
Table 9.
Xeroderma Pigmentosum: Notable Pathogenic Variants by Gene
Chapter Notes
Author Notes
NIH Study 99-C-0099. Examination of Clinical and Laboratory Abnormalities in Patients with Defective DNA Repair: Xeroderma Pigmentosum, Cockayne Syndrome, or Trichothiodystrophy is actively recruiting new patients for a study in Bethesda, MD. Click here for more information.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and National Human Genome Research Institute.
We wish to thank the many patients and families throughout the world who have participated in the XP protocols at the National Institutes of Health, National Cancer Institute.
Author History
John J DiGiovanna, MD (2011-present)
Kenneth H Kraemer, MD (2003-present)
Deborah Tamura, RN (2022-present)
Daniel J Wattendorf, MD; National Institutes of Health (2003-2008)
Revision History
- 24 March 2022 (ha) Comprehensive update posted live
- 29 September 2016 (ma) Comprehensive update posted live
- 13 February 2014 (me) Comprehensive update posted live
- 14 February 2013 (cd) Revision: changes in testing available for POLH, ERCC3, XPA, XPC, and DDB2
- 1 November 2012 (cd) Revision: testing for ERCC4 mutations available clinically; Figure 2 added
- 15 March 2012 (cd) Revision: sequence analysis available clinically for ERCC1 and ERCC3 and no longer available for DDB2
- 4 August 2011 (me) Comprehensive update posted live
- 22 April 2008 (me) Comprehensive update posted live
- 14 May 2007 (cd) Revision: sequence analysis clinically available for XPA and XPC
- 1 June 2006 (cd) Revision: confirmation of XPA and XPC mutations identified in a research lab clinically available
- 15 September 2005 (me) Comprehensive update posted live
- 24 February 2004 (kk) Revision: Molecular Genetics
- 1 October 2003 (kk) Revision: clinical testing no longer available
- 20 June 2003 (me) Review posted live
- 28 April 2003 (kk) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Xeroderma Pigmentosum" is in the public domain in the United States of America.
References
Published Guidelines / Consensus Statements
- Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori C, et al. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44:1087–96. [PubMed: 28771907]
- Tamura D, Kraemer KH, DiGiovanna JJ. Xeroderma pigmentosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, Coulson I, eds. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 4 ed. London, UK: Elsevier; 2014.
Literature Cited
- Ali JT, Mukasa Y, Coulson IH. Xeroderma pigmentosum: early diagnostic features and an adverse consequence of photoprotection. Clin Exp Dermatol. 2009;34:442–3. [PubMed: 19309384]
- Arlett CF, Plowman PN, Rogers PB, Parris CN, Abbaszadeh F, Green MH, McMillan TJ, Bush C, Foray N, Lehmann AR. Clinical and cellular ionizing radiation sensitivity in a patient with xeroderma pigmentosum. Br J Radiol. 2006;79:510–7. [PubMed: 16714754]
- Ben Rekaya M, Jerbi M, Messaoud O, Ben Brick AS, Zghal M, Mbarek C, Chadli-Debbiche A, Jones M, Mokni M, Boussen H, Boubaker MS, Fazaa B, Yacoub-Youssef H, Abdelhak S. Further evidence of mutational heterogeneity of the XPC gene in Tunisian families: a spectrum of private and ethnic specific mutations. Biomed Res Int. 2013;2013:316286. [PMC free article: PMC3741899] [PubMed: 23984341]
- Ben Rekaya M, Laroussi N, Messaoud O, Jones M, Jerbi M, Naouali C, Bouyacoub Y, Chargui M, Kefi R, Fazaa B, Boubaker MS, Boussen H, Mokni M, Abdelhak S, Zghal M, Khaled A, Yacoub-Youssef H. A founder large deletion mutation in xeroderma pigmentosum-Variant form in Tunisia: implication for molecular diagnosis and therapy. Biomed Res Int. 2014;2014:256245. [PMC free article: PMC4024419] [PubMed: 24877075]
- Ben Rekaya M, Messaoud O, Talmoudi F, Nouira S, Ouragini H, Amouri A, Boussen H, Boubaker S, Mokni M, Mokthar I, Abdelhak S, Zghal M. High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet. 2009;54:426–9. [PubMed: 19478817]
- Berneburg M, Kraemer KH. Xeroderma pigmentosum and other DNA repair-deficient photodermatoses. In: Lim H, Hönigsmann H, Hawk JLM, eds. Photodermatology. New York, NY: Informa Healthcare; 2007:239-50.
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, eds. The Genetic Basis of Human Cancer. 2 ed. New York, NY: McGraw-Hill; 2002:211-37.
- Boyle J, Ueda T, Oh KS, Imoto K, Tamura D, Jagdeo J, Khan SG, Nadem C, Digiovanna JJ, Kraemer KH. Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy. Hum Mutat. 2008;29:1194–208. [PMC free article: PMC3477783] [PubMed: 18470933]
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Pike KM, Raziuddin A, Plona TM, DiGiovanna JJ, Tucker MA, Kraemer KH. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–76. [PMC free article: PMC3235003] [PubMed: 21097776]
- Brooks BP, Thompson AH, Bishop RJ, Clayton JA, Chan CC, Tsilou ET, Zein WM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Iliff NT, Bradford P, Digiovanna JJ, Kraemer KH. Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology. 2013;120:1324–36. [PMC free article: PMC3702678] [PubMed: 23601806]
- Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, Stefanini M, Menefee E, Price VH, Queille S, Sarasin A, Bohnert E, Krutmann J, Davidson R, Kraemer KH, Lehmann AR. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet. 2001;10:2539–47. [PubMed: 11709541]
- Butt FM, Moshi JR, Owibingire S, Chindia ML. Xeroderma pigmentosum: a review and case series. J Craniomaxillofac Surg. 2010;38:534–7. [PubMed: 20346687]
- Calmels N, Greff G, Obringer C, Kempf N, Gasnier C, Tarabeux J, Miguet M, Baujat G, Bessis D, Bretones P, Cavau A, Digeon B, Doco-Fenzy M, Doray B, Feillet F, Gardeazabal J, Gener B, Julia S, Llano-Rivas I, Mazur A, Michot C, Renaldo-Robin F, Rossi M, Sabouraud P, Keren B, Depienne C, Muller J, Mandel JL, Laugel V. Uncommon nucleotide excision repair phenotypes revealed by targeted high-throughput sequencing. Orphanet J Rare Dis. 2016;11:26. [PMC free article: PMC4804614] [PubMed: 27004399]
- Chambon F, Osdoit S, Bagny K, Moro A, Nguyen J, Réguerre Y. Dramatic response to nivolumab in xeroderma pigmentosum skin tumor. Pediatr Blood Cancer. 2018:65. [PubMed: 28988442]
- Christen-Zaech S, Imoto K, Khan SG, Oh KS, Tamura D, Digiovanna JJ, Boyle J, Patronas NJ, Schiffmann R, Kraemer KH, Paller AS. Unexpected occurrence of xeroderma pigmentosum in an uncle and nephew. Arch Dermatol. 2009;145:1285–91. [PMC free article: PMC3472955] [PubMed: 19917958]
- Clement FC, Camenisch U, Fei J, Kaczmarek N, Mathieu N, Naegeli H. Dynamic two-stage mechanism of versatile DNA damage recognition by xeroderma pigmentosum group C protein. Mutat Res. 2010;685:21–8. [PubMed: 19686765]
- Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol. 2015;173:M85–97. [PMC free article: PMC4553126] [PubMed: 26130139]
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–96. [PMC free article: PMC3279615] [PubMed: 22217736]
- DiGiovanna JJ, Patronas N, Katz D, Abangan D, Kraemer KH. Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg. 1998;2:153–8. [PubMed: 9479081]
- Falik-Zaccai TC, Erel-Segal R, Horev L, Bitterman-Deutsch O, Koka S, Chaim S, Keren Z, Kalfon L, Gross B, Segal Z, Orgal S, Shoval Y, Slor H, Spivak G, Hanawalt PC. A novel XPD mutation in a compound heterozygote; the mutation in the second allele is present in three homozygous patients with mild sun sensitivity. Environ Mol Mutagen. 2012;53:505–14. [PubMed: 22826098]
- Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, Craythorne E, Morley AM, Lim R, Turner S, Henshaw T, Garrood I, Giunti P, Hedderly T, Abiona A, Naik H, Harrop G, McGibbon D, Jaspers NG, Botta E, Nardo T, Stefanini M, Young AR, Sarkany RP, Lehmann AR. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016;113:E1236–45. [PMC free article: PMC4780618] [PubMed: 26884178]
- Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A, Wijgers N, Jaspers NG, Raams A, Argentini M, Van der Spek PJ, Botta E, Stefanini M, Egly JM, Aebersold R, Hoeijmakers JH, Vermeulen W. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet. 2004;36:714–9. [PubMed: 15220921]
- Graham JM Jr, Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH, Busch D, Edersheim TG, Jaspers NG. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet. 2001;69:291–300. [PMC free article: PMC1235303] [PubMed: 11443545]
- Hadj-Rabia S, Oriot D, Soufir N, Dufresne H, Bourrat E, Mallet S, Poulhalon N, Ezzedine K, Grandchamp B, Taïeb A, Catteau B, Sarasin A, Bodemer C. Unexpected extradermatological findings in 31 patients with xeroderma pigmentosum type C. Br J Dermatol. 2013;168:1109–13. [PubMed: 23278166]
- Heller ER, Khan SG, Kuschal C, Tamura D, DiGiovanna JJ, Kraemer KH. Mutations in the TTDN1 gene are associated with a distinct trichothiodystrophy phenotype. J Invest Dermatol. 2015;135:734–41. [PMC free article: PMC4530629] [PubMed: 25290684]
- Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee DG, Kodama K, Mabuchi K, Kraemer KH, Land CE, Nakamura N. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601:171–8. [PubMed: 16905156]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci USA. 2004;101:15410–5. [PMC free article: PMC524447] [PubMed: 15486090]
- Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol. 2001;44:891–920. [PubMed: 11369901]
- Itoh T, Ono T, Yamaizumi M. A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat Res. 1994;314:233–48. [PubMed: 7513056]
- Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D, Kleijer WJ, Hoeijmakers JH, Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80:457–66. [PMC free article: PMC1821117] [PubMed: 17273966]
- Jerbi M, Ben Rekaya M, Naouali C, Jones M, Messaoud O, Tounsi H, Nagara M, Chargui M, Kefi R, Boussen H, Mokni M, Mrad R, Boubaker MS, Abdelhak S, Khaled A, Zghal M, Yacoub-Youssef H. Clinical, genealogical and molecular investigation of the xeroderma pigmentosum type C complementation group in Tunisia. Br J Dermatol. 2016;174:439–43. [PubMed: 26211814]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, Hirano A, Yamashita S, Greenblatt D, Nardo T, Stefanini M, McGibbon D, Sarkany R, Fassihi H, Takahashi Y, Nagayama Y, Mitsutake N, Lehmann AR, Ogi T. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92:807–19. [PMC free article: PMC3644632] [PubMed: 23623389]
- Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, Jaspers NG, Sarasin A, Stefanini M, Lehmann AR. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst). 2008;7:744–50. [PubMed: 18329345]
- Kouatcheu SD, Marko J, Tamura D, Khan SG, Lee CR, DiGiovanna JJ, Kraemer KH. Thyroid nodules in xeroderma pigmentosum patients: a feature of premature aging. J Endocrinol Invest. 2021;44:1475–82. [PMC free article: PMC8096868] [PubMed: 33155181]
- Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med. 1988;318:1633–7. [PubMed: 3287161]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–21. [PubMed: 8053698]
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. [PubMed: 3545087]
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145:1388–96. [PMC free article: PMC2288663] [PubMed: 17276014]
- Kraemer KH, Ruenger T. Genome instability, DNA repair, and cancer. In: Wolff K, Katz S, Goldsmith L, Gilchrist B, Leffell D, Paller A, eds. Fitzpatrick's Dermatology in General Medicine. 8 ed. New York, NY: McGraw-Hill; 2012:1227-39.
- Kraemer KH, Slor H. Xeroderma pigmentosum. Clin Dermatol. 1985;3:33–69. [PubMed: 3833325]
- Kuschal C, Botta E, Orioli D, Digiovanna JJ, Seneca S, Keymolen K, Tamura D, Heller E, Khan SG, Caligiuri G, Lanzafame M, Nardo T, Ricotti R, Peverali FA, Stephens R, Zhao Y, Lehmann AR, Baranello L, Levens D, Kraemer KH, Stefanini M. GTF2E2 mutations destabilize the general transcription factor complex TFIIE in individuals with DNA repair-proficient trichothiodystrophy. Am J Hum Genet. 2016;98:627–42. [PMC free article: PMC4833217] [PubMed: 26996949]
- Lai JP, Liu YC, Alimchandani M, Liu Q, Aung PP, Matsuda K, Lee CC, Tsokos M, Hewitt S, Rushing EJ, Tamura D, Levens DL, DiGiovanna JJ, Fine HA, Patronas N, Khan SG, Kleiner DE, Oberholtzer JC, Quezado MM, Kraemer KH. The influence of DNA repair on neurologic degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D). Acta Neuropathol Commun. 2013;1:4. [PMC free article: PMC3776212] [PubMed: 24252196]
- Lambert WC, Lambert MW. Development of effective skin cancer treatment and prevention in xeroderma pigmentosum. Photochem Photobiol. 2015;91:475–83. [PubMed: 25382223]
- Lehky TJ, Sackstein P, Tamura D, Quezado M, Wu T, Khan SG, Patronas NJ, Wiggs E, Brewer CC, DiGiovanna JJ, Kraemer KH. Differences in peripheral neuropathy in xeroderma pigmentosum complementation groups A and D as evaluated by nerve conduction studies. BMC Neurol. 2021;21:393. [PMC free article: PMC8501575] [PubMed: 34627174]
- Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E, DiGiovanna JJ. Characterization of tiger tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol. 2005;52:224–32. [PubMed: 15692466]
- Mahindra P, DiGiovanna JJ, Tamura D, Brahim JS, Hornyak TJ, Stern JB, Lee CC, Khan SG, Brooks BP, Smith JA, Driscoll BP, Montemarano AD, Sugarman K, Kraemer KH. Skin cancers, blindness, and anterior tongue mass in African brothers. J Am Acad Dermatol. 2008;59:881–6. [PMC free article: PMC2717011] [PubMed: 19119101]
- Martens MC, Emmert S, Boeckmann L. Xeroderma pigmentosum: gene variants and splice variants. Genes (Basel). 2021;12:1173. [PMC free article: PMC8391564] [PubMed: 34440347]
- Masaki T, Ono R., Tanioka M., Funasaka Y., Nagano T., Moriwaki S., Nishigori C. Four types of possible founder mutations are responsible for 87% of Japanese patients with Xeroderma pigmentosum variant type. J Dermatol Sci. 2008;52:144–8. [PubMed: 18703314]
- Meira LB, Graham JM Jr, Greenberg CR, Busch DB, Doughty AT, Ziffer DW, Coleman DM, Savre-Train I, Friedberg EC. Manitoba aboriginal kindred with original cerebro-ocular facial syndrome. Am J Hum Genet. 2000;66:1221–8. [PMC free article: PMC1288189] [PubMed: 10739753]
- Merideth M, Tamura D, Angra D, Khan SG, Ferrell J, Goldstein AM, DiGiovanna JJ, Kraemer KH. Reproductive health in xeroderma pigmentosum: features of premature aging. Obstet Gynecol. 2019;134:814–9. [PMC free article: PMC6768713] [PubMed: 31503159]
- Messaoud O, Ben Rekaya M, Kefi R, Chebel S, Boughammoura-Bouatay A, Bel Hadj Ali H, Gouider-Khouja N, Zili J, Frih-Ayed M, Mokhtar I, Abdelhak S, Zghal M. Identification of a primarily neurological phenotypic expression of xeroderma pigmentosum complementation group A in a Tunisian family. Br J Dermatol. 2010;162:883–6. [PubMed: 20199544]
- Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori C, et al. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44:1087–96. [PubMed: 28771907]
- Moslehi R, Signore C, Tamura D, Mills JL, Digiovanna JJ, Tucker MA, Troendle J, Ueda T, Boyle J, Khan SG, Oh KS, Goldstein AM, Kraemer KH. Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development. Clin Genet. 2010;77:365–73. [PMC free article: PMC3463936] [PubMed: 20002457]
- Munford V, Castro LP, Souto R, Lerner LK, Vilar JB, Quayle C, Asif H, Schuch AP, de Souza TA, Ienne S, Alves FIA, Moura LMS, Galante PAF, Camargo AA, Liboredo R, Pena SDJ, Sarasin A, Chaibub SC, Menck CFM. A genetic cluster of patients with variant xeroderma pigmentosum with two different founder mutations. Br J Dermatol. 2017;176:1270–8. [PubMed: 27664908]
- Nakano E, Masaki T, Kanda F, Ono R, Takeuchi S, Moriwaki S, Nishigori C. The present status of xeroderma pigmentosum in Japan and a tentative severity classification scale. Exp Dermatol. 2016;25 Suppl 3:28–33. [PubMed: 27539899]
- Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, Takahashi Y, Ohyama K, Ito K, Mishima H, Nomura M, Kinoshita A, Ono S, Takenaka K, Masuyama R, Kudo T, Slor H, Utani A, Tateishi S, Yamashita S, Stefanini M, Lehmann AR, Yoshiura K, Ogi T. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet. 2012;44:586–92. [PubMed: 22466610]
- Nardo T, Oneda R, Spivak G, Vaz B, Mortier L, Thomas P, Orioli D, Laugel V, Stary A, Hanawalt PC, Sarasin A, Stefanini M. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci USA. 2009;106:6209–14. [PMC free article: PMC2667150] [PubMed: 19329487]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst FTJ, Meinecke P, Kleijer WJ, Vijg J, Jaspers NGJ, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43. [PubMed: 17183314]
- Nikolaev S, Yurchenko AA, Sarasin A. Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J Rare Dis. 2022;17:104. [PMC free article: PMC8896305] [PubMed: 35246173]
- Oetjen KA, Levoska MA, Tamura D, Ito S, Douglas D, Khan SG, Calvo KR, Kraemer KH, DiGiovanna JJ. Predisposition to hematologic malignancies in patients with xeroderma pigmentosum. Haematologica. 2020;105:e144–e146. [PMC free article: PMC7109711] [PubMed: 31439674]
- Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A, Friedmann PS, Emmert S, Gratchev A, Lachlan K, Lucassan A, Baker CC, Kraemer KH. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27:1092–103. [PubMed: 16947863]
- Ohto T, Iwasaki N, Okubo H, Shin K, Matsui A. Life-threatening vocal cord paralysis in a patient with group A xeroderma pigmentosum. Pediatr Neurol. 2004;30:222–4. [PubMed: 15033209]
- Opletalova K, Bourillon A, Yang W, Pouvelle C, Armier J, Despras E, Ludovic M, Mateus C, Robert C, Kannouche P, Soufir N, Sarasin A. Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations. Hum Mutat. 2014;35:117–28. [PubMed: 24130121]
- Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–9. [PMC free article: PMC4459578] [PubMed: 11185579]
- Reichrath J. Sunlight, skin cancer and vitamin D: What are the conclusions of recent findings that protection against solar ultraviolet (UV) radiation causes 25-hydroxyvitamin D deficiency in solid organ-transplant recipients, xeroderma pigmentosum, and other risk groups? J Steroid Biochem Mol Biol. 2007;103:664–7. [PubMed: 17204418]
- Rizza ERH, DiGiovanna JJ, Khan SG, Tamura D, Jeskey JD, Kraemer KH. Xeroderma Pigmentosum: A Model for Human Premature Aging. J Invest Dermatol. 2021;141:976–84. [PMC free article: PMC7987754] [PubMed: 33436302]
- Ruenger T, DiGiovanna J, Kraemer KH. Hereditary diseases of genome instability and DNA repair. In: Wolff K, Katz S, Goldsmith L, Gilchrist B, Leffell D, Paller A, eds. Fitzpatrick's Dermatology in General Medicine. 8 ed. New York, NY: McGraw-Hill; 2012:1654-71.
- Salomon G, Maza A, Boulinguez S, Paul C, Lamant L, Tournier E, Mazereeuw-Hautier J, Meyer N. Efficacy of anti-programmed cell death-1 immunotherapy for skin carcinomas and melanoma metastases in a patient with xeroderma pigmentosum. Br J Dermatol. 2018;178:1199–203. [PubMed: 29274233]
- Sarasin A, Munier P, Cartault F. How history and geography may explain the distribution in the Comorian archipelago of a novel mutation in DNA repair-deficient xeroderma pigmentosum patients. Genet Mol Biol. 2019a;43:e20190046. [PMC free article: PMC7198018] [PubMed: 31930276]
- Sarasin A, Quentin S, Droin N, Sahbatou M, Saada V, Auger N, Boursin Y, Dessen P, Raimbault A, Asnafi V, Schmutz JL, Taieb A, Menck CFM, Rosselli F, La Rochelle LD, Robert C, Sicre de Fontbrune F, Sebert M, Leblanc T, Kannouche P, De Botton S, Solary E, Soulier J. Familial predisposition to TP53/complex karyotype MDS and leukemia in DNA repair-deficient xeroderma pigmentosum. Blood. 2019b;133:2718–24. [PMC free article: PMC6610036] [PubMed: 30914417]
- Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M, Vermeulen W, Marteijn JA. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44:598–602. [PubMed: 22466611]
- Sethi M, Lehmann AR, Fawcett H, Stefanini M, Jaspers N, Mullard K, Turner S, Robson A, McGibbon D, Sarkany R, Fassihi H. Patients with xeroderma pigmentosum complementation groups C, E and V do not have abnormal sunburn reactions. Br J Dermatol. 2013;169:1279–87. [PubMed: 23889214]
- Shanbhag NM, Geschwind MD, DiGiovanna JJ, Groden C, Godfrey R, Yousefzadeh MJ, Wade EA, Niedernhofer LJ, Malicdan MCV, Kraemer KH, Gahl WA, Toro C. Neurodegeneration as the presenting symptom in 2 adults with xeroderma pigmentosum complementation group F. Neurol Genet. 2018;4:e240. [PMC free article: PMC5994703] [PubMed: 29892709]
- Sollitto RB, Kraemer KH, DiGiovanna JJ. Normal vitamin D levels can be maintained despite rigorous photoprotection: Six years' experience with xeroderma pigmentosum. J Am Acad Dermatol. 1997;37:942–7. [PubMed: 9418761]
- Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A, Armier J, Pham D, Khadir K, Roume J, Hadj-Rabia S, Bouadjar B, Taieb A, de Verneuil H, Benchiki H, Grandchamp B, Sarasin A. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol. 2010;130:1537–42. [PubMed: 20054342]
- Stefanini M, Kraemer KHK. Xeroderma pigmentosum. In: Ruggieri M, Pascual-Castroviejo I, Di Rocco C, eds. Neurocutaneous Diseases. New York, NY: Springer; 2008:771-92.
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Sugasawa K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res. 2010;685:29–37. [PubMed: 19682467]
- Tamhankar PM, Iyer SV, Ravindran S, Gupta N, Kabra M, Nayak C, Kura M, Sanghavi S, Joshi R, Chennuri VS, Khopkar U. Clinical profile and mutation analysis of xeroderma pigmentosum in Indian patients. Indian J Dermatol Venereol Leprol. 2015;81:16–22. [PubMed: 25566891]
- Tamura D, Khan SG, Merideth M, DiGiovanna JJ, Tucker MA, Goldstein AM, Oh K-S, Ueda T, Boyle J, Sarihan M, Kraemer KH. Effect of mutations in XPD (ERCC2) on pregnancy and prenatal development in mothers of patients with trichothiodystrophy or xeroderma pigmentosum. Eur J Hum Genet. 2012;20:1308–10. [PMC free article: PMC3499748] [PubMed: 22617342]
- Tamura D, Merideth M, DiGiovanna JJ, Zhou X, Tucker MA, Goldstein AM, Brooks BP, Khan SG, Oh KS, Ueda T, Boyle J, Moslehi R, Kraemer KH. High-risk pregnancy and neonatal complications in the DNA repair and transcription disorder trichothiodystrophy: report of 27 affected pregnancies. Prenat Diagn. 2011;31:1046–53. [PMC free article: PMC3266696] [PubMed: 21800331]
- van Steeg H, Kraemer KH. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol Med Today. 1999;5:86–94. [PubMed: 10200950]
- Wilson BT, Lochan A, Stark Z, Sutton RE. Novel missense mutations in a conserved loop between ERCC6 (CSB) helicase motifs V and VI: Insights into Cockayne syndrome. Am J Med Genet A. 2016;170:773–6. [PubMed: 26749132]
- Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T, Yasui A, Tanaka K. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat Genet. 2012;44:593–7. [PubMed: 22466612]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: June 20, 2003; Last Update: March 24, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Kraemer KH, DiGiovanna JJ, Tamura D. Xeroderma Pigmentosum. 2003 Jun 20 [Updated 2022 Mar 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

