

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction
Jason Manka, Richard N. Daniels, Eric Dawson, J. Scott Daniels, Noel Southall, Ajit Jadhav, Wei Zheng, Chris Austin, Jolanta Grembecka, Tomasz Cierpicki, Craig W. Lindsley, and Shaun R. Stauffer.
Author Information and Affiliations .
.Received: March 31, 2011; Last Update: February 28, 2013.
A series of lead structures identified from a High Throughput Screen (HTS) targeting the Menin-Mixed Lineage Leukemia (MLL) protein-protein interaction are reported. Two chemical series have been prosecuted to date and one piperidine series was identified to have tractable and rapidly response Structure Activity Relationship (SAR) affording inhibitors of the Menin-MLL interaction with sub-micromolar inhibitory activity. Moreover, preliminary data suggests these compounds display activity in cellular systems relevant to understanding the Menin-MLL pathway and the disease progression. SAR and characterization of the declared probe ML227 from this effort are described.
Assigned Assay Grant #: MH084875
Screening Center Name & PI: NIH Chemical Genomics Center, Chris Austin
Chemistry Center Name & PI: Vanderbilt Specialized Chemistry Center, Craig Lindsley
Assay Submitter & Institution: Jolanta Grembecka, University of Michigan
PubChem Summary Bioassay Identifier (AID): 2076
Probe Structure & Characteristics
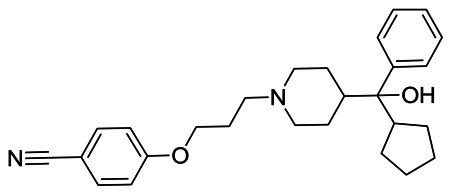
4-(3-(4-(cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile, MW=418.57, cLogP=5.19, tPSA=56.5 Å2
| CID/ML# | Target Name | IC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 46926631/ML227 | Menin-MLL | 883 nM [SID 99432383, AID 2076 | None | NA | NA | NA |
Recommendations for Scientific Use of the Probe
This probe (ML227, CID 46926631, SID 99432383) represents the first Menin-Mixed Lineage Leukemia (MLL) small molecule protein-protein interaction inhibitor with sub-micromolar inhibitory activity. This probe will be used by the research community to further elucidate the importance of the menin-MLL interaction in MLL-mediated leukemogenesis and offers a potential drug discovery path to develop small molecules to treat acute lymphoid and myeloid leukemias with MLL rearrangements Translocations of MLL result in acute leukemias with poor prognosis and development of novel therapeutic strategies is highly desired. Leukemogenic activity of MLL fusion proteins is dependent of their interactions with menin, validating the importance of this interaction as a potential drug target for leukemia [13,14]. Currently, peptide fragments have been identified which are reported to be efficacious in disrupting the Menin-MLL interactions in vitro and in cells [14, 22]. Although development of small peptide disruptors is generally useful for mapping features of the active binding surface and as probes for screening purposes, these tools are typically not viable as lead starting points for small molecule drug discovery. In light of the compelling evidence to date identifying the Menin-MLL fusion protein interaction as a rate limiting interaction necessary for leukemic cell proliferation and blockage of hematopoietic differentiation, the community is earnestly searching for small molecules to disrupt and block the Menin-MLL mediated pathway [22] In addition to utilizing a probe molecule to further validate the molecular target, we are focused on identifying good probe compounds to serve as leads for further lead development within the context of the extended probe mechanism. Such compounds might result in novel targeted therapies for MLL-associated acute leukemias, and could also be used a chemical probes to study the biology of MLL and MLL fusion protein mediated leukemogenesis.
1. Introduction
Specific AIM: Identification of functional inhibitors targeting Menin and blocking the Menin-MLL interaction with low micromolar to submicromolar potency, to be used as probes to understand the biological consequence of inhibiting this interaction and its impact on leukemia development. Ultimately, this work may result in the development of novel drugs for effective treatment of MLL acute leukemias.
Significance: Chromosomal translocations involving the mixed lineage leukemia (MLL) gene result in human acute myeloid and lymphoid leukemias, affecting both children and adults (1,2). Fusion of MLL with one of 60 partner genes forms chimeric oncogenes encoding MLL fusion proteins, which results in enhanced proliferation and blockage of blood cell differentiation ultimately leading to the development of acute leukemia (3). Translocations of MLL are particularly prevalent in infants with AML (acute myeloid leukemia) and ALL (acute lymphoblastic leukemia), and constitute up to 80% of all infant acute leukemia cases (4). Patients with leukemias harboring MLL translocations have a very poor prognosis using available therapies (20 % event-free survival at 3 years), and it is clear that novel targeted therapies are urgently needed to treat these leukemias (3,5).
MLL is an important regulator of Hox gene expression, which is required for normal hematopoiesis (10). Disruption of MLL by chromosomal translocations upregulates expression of Hox genes, including Hoxa7, Hoxa9 and the Hox cofactor Meis1, resulting in blockage of hematopoietic differentiation that leads to leukemia (11). MLL is involved in a complex network of interactions with multiple proteins, including menin (12,13). Importantly, the direct interaction with menin is critical for the oncogenic function of MLL fusion proteins (13–15). Menin is a highly specific partner for MLL proteins and is an essential component of the MLL SET1-like histone methyltransferase (HMT) complex (12,16). The Menin-MLL interaction is required to regulate expression of MLL target genes, including Hoxa7, Hoxa9, Hoxa10, Hoxc7 and Meis1 (12–15). Menin is a 67 kDa tumor suppressor protein encoded by the Men1 (Multiple Endocrine Neoplasia I) gene (17), which directly controls cell growth in selected organs, including parathyroid, pancreatic islets, and the pituitary gland (18). In leukemias, menin functions as an essential oncogenic co-factor of MLL fusion proteins (13). Since menin binds to the N-terminus of MLL, this interaction is preserved among wild-type MLL and all MLL-fusion proteins (13,14). Mutations within the N-terminus of MLL fusions disrupt its association with menin and abolish its oncogenic properties in vitro and in vivo (13–15). Furthermore, the expression of a dominant-negative polypeptide corresponding to the amino terminal MLL sequence inhibits growth of MLL-AF9 transformed bone marrow cells (14). Overall, the menin interaction with MLL is critical for the oncogenic activity of MLL fusions, validating the menin-MLL interaction as a potential target for molecular therapy (13,14). Recent findings strongly suggest that MLL fusion proteins require the co-expression of wild-type MLL to induce leukemia (19). Therefore, inhibition of the association of menin with both MLL and MLL fusions by small molecule inhibitors might result in new therapeutic agents for MLL-associated leukemias. In addition, such an inhibitor will be highly valuable to the research community in dissecting the role of menin-MLL interactions in MLL-mediated leukemogenesis and for the function of menin as a tumor suppressor.
Rationale: No legitimate non-covalent small molecule inhibitors targeting the menin-MLL interaction have been described to date. Therefore, inhibition of the association of menin with both MLL and MLL fusions by small molecule inhibitors are needed to test the Menin function as an oncogenic cofactor in MLL leukemias as a means for new therapeutic intervention. Menin is now a well characterized protein and furthermore peptide-based fragment inhibitors of the Menin-MLL interaction have been established and have been shown to inhibit growth of MLL-AF9 transformed bone marrow cells, down regulate expression of cancer-related target genes and induce hematopoietic differentiation (14). We expect to achieve the same effect with small molecule inhibitors targeting the menin-MLL interaction. In summary, characterization of this target has matured and now provides an informed basis and position to interpret and prioritize hits from a comprehensive screen.
2. Materials and Methods
2.1. Assays
PubChem Primary FP Assay Description using Fluorescein: The purpose of this biochemical in vitro fluorescence polarization assay is to screen for inhibitors of the menin-MLL (Mixed Lineage Leukemia) interaction which might result in identification of lead scaffolds for the development of effective drug treatments for MLL-related acute leukemias (AID 1768, Summary 2076). In this assay, a fluorescein labeled MLL-derived peptide (FLSN_MLL) and full length menin were used. Binding of FLSN_MLL peptide to menin is reflected by a substantial increase in fluorescence polarization signal. Inhibition of this interaction by small molecules relieves FLSN_MLL peptide from menin, resulting in increased molecular tumbling of the free peptide in solution, which is reflected by a significant decrease in fluorescence polarization (FP) signal. The decrease in FP signal has been used as a measure of inhibition of the menin-MLL interaction by HTS compounds.
Other Primary Assay FP Description using Texas Red Dye: This purpose of this assay is to operate as a secondary FP screen for hit validation. This assay utilizes a Texas Red tag on the MLL peptide (AID 1766) instead of Fluoroscein which is used in the primary assay AID 1768. This assay is not used during SAR for the hit-to-lead or lead optimization phase of the project.
Secondary HTRF Assay Description: This purpose of this assay is to provide confidence in hit selection in addition to the two primary FP assays (AID 2278). This assay utilizes HTRF (commercial TR-FRET) using a His-tagged menin and biotin-labeled MLL peptide.
Secondary NMR Assay Description: This purpose of this assay is to provide further confidence in hit selection in addition to HTRF (AID 2781). This assay utilizes Saturation Transfer Difference (STD) NMR spectroscopy to verify direct binding of compounds to menin and their inhibition of menin interaction with MLL.
2.2. Probe Chemical Characterization
Synthetic procedure and spectral data for ML227 (CID 46926631, SID 99432383)

Route A: ML227 Synthesis
4-RS-(3-(4-(cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile
Probe compound ML227 (CID 46926631) was prepared according to the above scheme and provided the following characterization data: LC-MS (>98%) m/z = 419.3 [M+H], 1H NMR (400 MHz, CDCl3) δ 7.54 (2H, d, J=8.8 Hz), 7.37 (2H, d, J=7.6 Hz), 7.31 (2H, t, J=7.6 Hz), 7.22 (1H, t, J=7.2 Hz), 6.89 (2H, d, J=8.8 Hz), 4.02 (2H, t, J=6 Hz), 3.12-3.00 (2H, m), 2.74-2.62 (3H, m), 2.09-1.95 (4H, m), 1.80-1.60 (4H, m), 1.58-1.40 (7H, m), 1.25-1.24 (1H, m), 1.11-1.09 (1H, m).
Note, the final Grignard addition described in Route A proceeds in low yield (<10%, see below). Route B, shown below, has recently been established as an alternative route. Route B precedes via a cyclopentenyl Grignard addition, followed reduction of the double bond. Preliminary results indicate this strategy provides a superior yield of final compound. Although route A readily allows small quantities of probe compound (50–100 mg), route B is recommended for long-term supply in greater than 100 mg quantities.
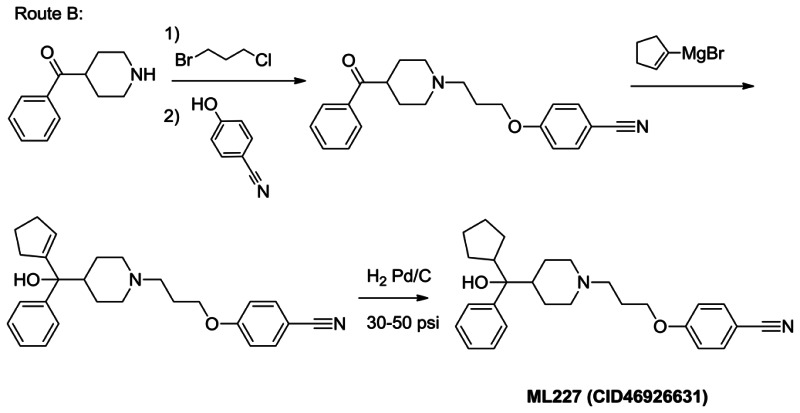
Route B: ML227 Synthesis
In an attempt to distinguish primary and ancillary activity of the individual enantiomers of ML227 a chiral Supercritical Fluid Chromatography (SFC) separation was performed (biological and DMPK characterization of the enantiomers of ML227 is described below under Results Section 3).
Separation of enantiomers was readily accomplished using supercritical fluid chromatography. The column was an IA (UV 250 nm, 10 × 250 mm, Chiral Technologies), eluent 55% EtOH w/ 0.1% diethylamine in CO2 (15mL/min).

Figure 1Analytical SFC chromatogram of racemic mixture prior to separation
Solubility. Solubility in PBS at pH 7.4 was determined to be 6.3 μM or 3 μg/mL. ML227 shows good solubility up to 40 mM DMSO which is currently used for original stocks of novel compounds shipped to the assay provider for testing.
Stability. Stability was determined for ML227 in PBS buffer at room temperature. After 1 hour, the percent of parent compound remaining was 46%, indicating significant apparent decomposition after exposure to PBS. In light of the very low solubility in PBS (< 10 μM) it must be recognized that a component of the apparent loss of material is perhaps due to precipitation of compound after prolonged storage in aqueous buffer. Further evaluation and sampling by LC/MS after exposure in aqueous-methanol mixtures does not indicate a significant chemical decomposition after storage overnight (95% intact).
In addition, ML227 was subjected to glutathione (GSH) incubation over 1h in order to establish if the probe molecule was free from formation of covalent adducts. No GSH adducts of ML227 were detected from these incubations (>98.9% intact, see Figure 2).
Figure 2
Ion extraction chromatogram showing trace of ML227 in HRMS scan of T=60 min sample after GSH incubation.
Compounds added to the SMR collection (MLS#s): MLS003431866 (ML227, CID 46926631, 22 mg), MLS003431861, MLS003431862, MLS003431863, MLS003431864, MLS003431865.
2.3. Probe Preparation (Route A)
4-(3-(4-Benzoylpiperidin-1-yl)propoxy)benzonitrile. 4-Benzoylpiperdine hydrobromide (2.70 g, 10 mmol) was combined with K2CO3 (6.9 g, 50 mmol) in DMF (~40 mL), followed by 1-bromo-3-chloropropane (1.55g, 10 mmol). The reaction progress was monitored by LC-MS and upon completion of the reaction (~2 h) 4-cyanophenol (1.3 g, 11mmol) was added and the reaction allowed to stir overnight. The mixture was poured onto water and extracted with ethyl acetate, washed with brine (2x) and dried over Na2SO4. The volatiles were removed under reduced pressure using a rotovap and the crude mixture purified on silica gel (0–100% EtOAc in hexane) to give 2.71g (78% yield over 2 steps) of the title intermediate compound: LC/MS [M+H] = 349.2.
4-(3-(4-(Cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile. 4-(3-(4-Benzoylpiperidin-1-yl)propoxy)benzonitrile (1.4 g, 4 mmol) was dissolved in dry THF (~25 mL), heated to 60° C and a solution of cyclopentylmagnesium chloride (4 mL, 2M) was added and the reaction stirred for 1 h. The reaction was quenched (sat. NH4Cl) and extracted into EtOAc (2x). The organic layers were dried over Na2SO4 and the volatiles removed under reduced pressure. The crude residue was purified on silica gel (0–4% MeOH in CH2Cl2). The combined fractions were further purified by reverse phase chromatography to give 117 mg (7% yield) of analytically pure title compound ML227.
3. Results
The primary HTS campaign included approximately 300K compounds which were screened at 60, 12.5, 2.5 and 0.5 μM. See summary Figure 3. The results from the HTS campaign were highly successful and optimization was performed on two series to date leading in one case to the probe molecule described herein.
Figure 3
Summary of HTS Campaign and Probe Optimization.
3.1. Summary of Screening Results
The primary Menin-MLL screen of 300,497 compounds generated 81 strong hits with greater than 30% inhibition in both channels from the assay and modest effects on total fluorescence. Several hundred other compounds have a less clear interpretation of activity from the primary screen. A Z-prime of 0.81 and a hit-rate of 0.27% were noted. 343 compounds were ordered from the Molecular Libraries Small Molecule Repository (MLSMR) and were tested for confirmation screen. This second screen produced 64 active compounds with significant concentration-responses. A subset of these actives were further validated using Saturation Transfer Difference (STD) NMR experiments and competition STD. STD allows for the verification of direct binding of compounds to menin and their competition with MLL peptide for binding to menin. This technique is the most direct measure of inhibiting the menin-MLL interaction by a small molecule.
Summary of Hit Scaffolds and Optimization Plan:Figure 4 summarizes the seven lead scaffolds identified after Saturation-Transfer Difference Nuclear Magnetic Resonance (STD NMR) verification, including information on the ligand efficiencies and inhibitory activity for representative compounds.

Figure 4
Lead Scaffolds Confirmed by STD NMR.
For the first cycle of optimization, Scaffolds 2 and 6 were selected (Figure 4 and 5) as high priority. Scaffold 3 is a quaternary amine and a known covalent modifier and therefore was dropped from further consideration. Scaffold 5 was also deprioritized in light of the weak STD, high MW and limited tractability to prepare analogs around the cyclic amidine core. Scaffolds 1, 7 and 4, in order of priority, are considered attractive backup leads of potential interest and are currently planned for a future effort pending further discussion and plans beyond ML227 (Scaffold 6) series.
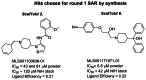
Figure 5
Scaffolds Selected for 1st Optimization Cycle.
Scaffold 2 Chemical Optimization and SAR Summary: In the aminopyrazole series, Scaffold 2, initial chemistry focused on modification of the piperidine N-alkyl substituent and the benzamide portion. Thirty-two compounds were synthesized and tested. No compounds were shown to demonstrate improved activity over the lead MLS001100636, although close analogs, including the saturated cyclohexyl derivative SID 85286042, appear to be nearly equipotent to the lead compound illustrating the importance of the size of the lipophilic carbocycle ring. Within the benzamide library, replacement of the 2-methoxy with the parent phenyl results in 4× loss of activity. Alternatively, a halogen substitution at the 4-position enhances activity similar to that found in MLS001100636. Further optimization of Scaffold 2 is on hold pending key post-probe plans around Scaffold 6. Long term a 2-MeO, 4-Cl di-substituted substitution pattern is planned to see if the independent potency enhancements will be additive. The synthetic plan and SAR highlights described are summarized in Figure 6.

Figure 6
Summary of Synthesis Plan and SAR highlights to date from Scaffold 2.
Scaffold 6 Chemical Optimization and Control Compounds: MLS001171971 was confirmed by STD NMR studies to be a relatively potent lead compound. As shown in Figure 7, the strategy for Scaffold 6 involved initially preparing a number of singletons to understand the importance of structural features which would synthetically dictate our ability to rapidly assess SAR. The key features being: 1) central hydrophilic OH group, 2) the three carbon aliphatic linker and 3) the tertiary carbinol. We first opted to tackle the linker and prepared the des-hydroxyl (CID 44543700) and R (CID 44543701) and S-enantiomers (CID 44543702) of the MLS001171971. From the initial studies acceptable variability in IC50 was found between the racemate MLS compound and the preferring R-ent CID 44543701. The S-ent CID 44543702 appears to be less tolerated (5–10x) with an IC50 of 45 μM. Interestingly, the des-hydroxyl compound CID 44543700 was only a modest 2–3× less active, suggesting that the central hydroxyl is not essential for activity.

Figure 7
1st Round Control Compounds for Scaffold 6.
We next turned to the next question (Figure 7) and examined the importance of both linker length and the position of the ether oxygen. As illustrated in Figure 7 from CID 46172912 and CID 46172901, a linker which is one carbon shorter or longer resulted in a deleterious effect on inhibitory activity. Furthermore, the isomeric analog CID 46172913, which places the ether oxygen in the benzylic position, results in a nearly complete loss of inhibition as well. This isomeric ether is predicted to have a high propensity to exist in a gauche conformation due to its β–proximity to the piperidine nitrogen (20). It’s not entirely clear if the loss in activity is purely driven by a conformational effect or due to loss in a critical Menin binding site interaction involving the ether oxygen. Additional analogs will be needed to further test this hypothesis; however, in light of the strong conformation effect of the gauche interaction (>20 kcal/mol) a conformational influence appears more likely. Lastly, to address the importance of the remaining hydroxyl we prepared CID 46172895 and CID 14407069, both of which were weak to inactive in their ability to disrupt the Menin-MLL interaction. Importantly, the control compounds presented herein allowed chemical space to be explored in other areas of the molecule in a more rapid fashion using fewer chemical steps (vide infra).
Scaffold 6 Optimization and SAR of the peripheral benzonitrile: After establishing that the central hydroxyl was not essential for activity, a rapid library of ethers was pursued using commercially available phenols and the common chloride intermediate 1 prepared from 1-bromo-3-chloropropane and the commercially available piperidine according to Scheme 1 below.
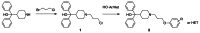
Scheme 1
Library of Scaffold 6 Ethers.
In addition, a small set of focused 4-cyano replacements were prepared, two of which utilized a sequence involving bromide CID 46926617 (entry 25, SAR Table 2); first conversion to azide CID 46926641 (entry 30, SAR Table 2) and subsequent tetrazole and triazole formation, to give CID 46926643 and CID 46926624 respectively (entry 31 and 28, SAR Table 2). In addition to their potential as hydrogen bond acceptors to replace the nitrile, we had hoped that such heterocycles would enhance solubility (Scheme not shown). To date, we have prepared and tested 33 direct analogs (SAR Table 2, section 3.4) of the des hydroxyl CID 44543700 (IC50 = 16 μM, Figure 7) benzonitrile compound. Among the various analogs evaluated, negatively charged groups were not well tolerated. Neutral polar or weakly basic charged groups, in particular the amino methyl derivative (CID 46926645, entry 32) and the 1,2,3-triazole derivative (CID 46926624, entry 28), had similar activity to the reference compound, 7–30 μM. A summary of the SAR highlights is shown below (Figure 8).
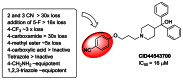
Figure 8
SAR Highlights from Benzonitrile Replacements (SAR Table 2).
Scaffold 6 Optimization and SAR of the peripheral diphenyl carbinol: Evaluation of the diphenyl carbinol SAR was initiated by reacting the diphenyl ketone precursor 3 with various nucleophiles to give analogs 4 (Scheme 2). To date, we have examined 13 various commercial Grignard reagents and Ruppert’s reagent to introduce a trifluoromethyl group (SAR Table 3, section 3.4).
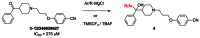
Scheme 2
Library of Scaffold 6 Carbinols.
Library 4 was highly successful in affording compounds with robust and orderly SAR. The highlights from this campaign are summarized in Figure 9. Reduction of the ketone to give alcohol CID 46926611 (R = H, entry 1) established the requirement for a bulky group to retain activity. Small acyclic aliphatic groups, such as a methyl group (entry 2) were equally weak at inhibiting the menin-MLL interaction (>200 μM). Addition of a n-butyl or branched i-propyl group (entry 3 and 4) were far more effective, in the case of the i-propyl analog achieving activity below 10 μM. A trifluoromethyl group, a common more metabolically stable i-propyl isotere, was incorporated to give a 31 μM inhibitor (entry 5). Two para substituted aromatics were tested and demonstrated a significant loss in activity, although tolerance for electron withdrawing groups was observed for the para-fluorine analog (net ~5× loss vs. H). More success was achieved through examination of cyclic aliphatic groups as phenyl replacements with the order of increasing activity being cyclopropyl < cyclobutyl < cyclohexyl < cyclopentyl. A clear relationship with respect to ring size was evident with cyclopentyl being optimal, demonstrating sub-micromolar disruption of the menin-MLL interaction for the first time (entry 8, CID 46926631 IC50 = 880 nM). CID 46926633 would go on to become ML227 however not without first carefully examining the enantiomers of CID 46926631. Since CID 46926631 is a mixture of R and S stereoisomers it is entirely conceivable that the mode of menin-MLL inhibition could be selective or preferring for one of the stereoisomers. Secondly, in order to extend probe impact it was also anticipated that the enantiomers may have differential profiles in terms of ancillary pharmacology (Ricerca) as well as DMPK properties (protein binding and metabolic stability). For these reasons, we pursued a chiral separation of the enantiomers via SFC (chromatogram and conditions described in Section 2.2) with the goal being to evaluate primary activity first.
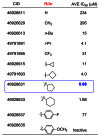
Figure 9
Carbinol Library Highlights.
Separation and Evaluation ML227 Enantiomers: SFC separation of CID 46926631 readily afforded the first and second eluting isomers designated as CID 49791889 and CID 49791890 (SAR Table 3 and Figure 10). Interestingly there was no significant difference in their primary activity. Similarly, we separated the R and S enantiomers of the cyclohexyl analog CID 46926633 and found little difference between the resulting pure stereoisomers (SAR Table 3 entries 10 and 11) in terms of their activity at disrupting the menin-MLL interaction.
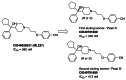
Figure 10
Enantiomers of ML227.
In light of the excellent inhibition observed for CID 46926631 as a PPI disruptor and the lack of differentiation between the enantiomers in the primary assay, we opted to submit the single enantiomers to both Ricerca and a tier one DMPK panel (VSCC in-house: plasma protein binding, intrinsic metabolic clearance and CYP 4-in-1 cocktail) in order to attempt to establish a possible preference for one of the stereoisomers between CID 49791889 and CID 49791890. The results of these efforts are summarized in Figure 11. Unfortunately, we saw no major differences in fraction unbound in rat plasma and both enantiomers were oxidatively metabolized quite rapidly in both human and rat microsomes (predicted hepatic clearance near liver blood flow for both species); indicating the potential in vivo utility of ML227 to be severely limited. More disappointing, but not entirely unexpected was the fact these compounds engage a number of off-target activities based upon competitive radioligand binding assays performed in Ricerca’s Lead Profiler screen (68 GPCRs, ion channels and transporters screened at 10 μM). In light of the profiles and reasons discussed above the team agreed that there is no advantage to declare one of the single stereoisomers of CID 46926631 as the probe at this time. Therefore, the racemic mixture CID 46926631 was nominated as the first probe compound.

Figure 11
Ancillary and DMPK Profiles of ML227 Enantiomers.
The calculated physical properties summarized in Table 1 for this menin-MLL probe molecule were generated using TRIPOS software. Also included in Table 1 are the averages from the MDDR database of compounds both entering Phase I and launched drugs. While some of the molecular properties of ML227, such as MW and HBA/HBD, are within range of Phase I compounds, the predicted solubility (LogS) and lipophilicity (cLogP) are clearly areas for improvement. What is particularly gratifying is the fact that typical PPI inhibitors published to date are substantially higher in MW versus ML227. Thus, with the current level of inhibition for ML227 and modest MW, the opportunity to improve the physiochemical properties while retaining good activity appears promising.
Table 1
Calculated Property Comparison with MDDR Compounds.
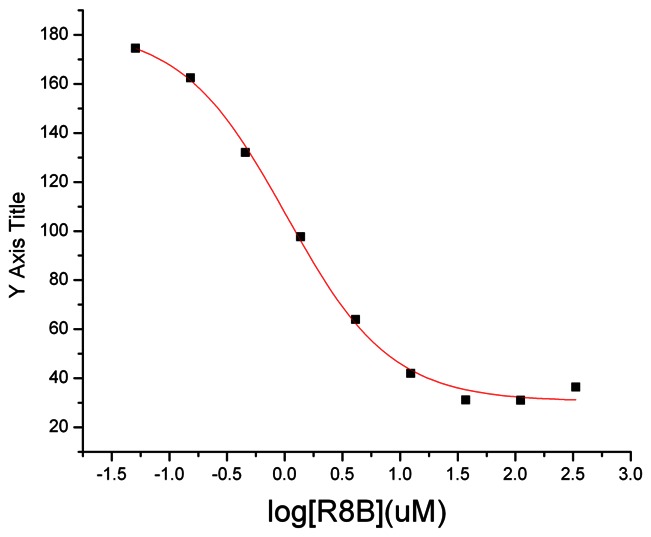
3.2. Dose Response Curves for Probe ML227 (barcode tested R8B)
3.3. Scaffold/Moiety Chemical Liabilities
There are no known chemical liabilities associated with ML227 (CID 46926631)
3.4. SAR Tables
Table 2SAR of Scaffold 6 Benzonitrile Replacements (<100 μM highlighted)
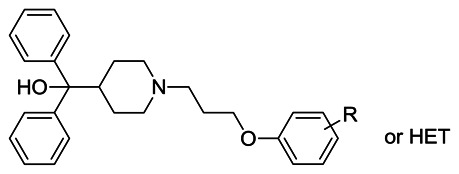 | |||||
|---|---|---|---|---|---|
| Entry | Ar/Het R- group | VU ID | CID | SID | AVE IC50 (μM, n of 2) |
| 1 | 2-CF3 | VU0413235 | 44907112 | 90744370 | 4.70E-04 |
| 2 | 4-CF3 | VU0413236 | 44907113 | 90744371 | 5.20E-05 |
| 3 | 3,5-diCH3 | VU0413237 | 44907114 | 90744372 | 4.81E-04 |
| 4 | 3-CN | VU0413238 | 44907115 | 90744373 | 9.24E-04 |
| 5 | 3-CF3 | VU0413239 | 44907116 | 90744374 | 6.35E-04 |
| 6 | 4-OCF3 | VU0413240 | 44907117 | 90744375 | 9.80E-05 |
| 7 | 3,5-diF | VU0413241 | 44907118 | 90744376 | 5.63E-04 |
| 8 | 3-OCF3 | VU0413242 | 44907119 | 90744377 | 5.46E-04 |
| 9 | 4-Cl | VU0413243 | 44907120 | 90744378 | 9.60E-05 |
| 10 | 4-CN, 5-F | VU0413244 | 44907121 | 90744379 | 2.65E-05 |
| 11 | 2-Cl | VU0413245 | 44902122 | 90744380 | 1.79E-04 |
| 12 | 3-F | VU0413246 | 44907123 | 90744381 | 4.44E-04 |
| 13 | 4-F | VU0413247 | 44907124 | 90744382 | 1.84E-04 |
| 14 | 2-CN | VU0413248 | 44907125 | 90744383 | 3.80E-04 |
| 15 | 2-OCH3 | VU0413249 | 44907126 | 90744384 | 9.07E-04 |
| 16 | 3-OCH3 | VU0413250 | 44907127 | 90744385 | 6.01E-04 |
| 17 | 2-pyridyl | VU0413251 | 44907128 | 90744386 | 1.50E-03 |
| 18 | 3-pyridyl | VU0413252 | 44907129 | 90744387 | 1.12E-03 |
| 19 | 2-CH3 | VU0413253 | 44907130 | 90744389 | 3.19E-04 |
| 20 | 3-CH3 | VU0413254 | 44907131 | 90744390 | 6.18E-04 |
| 21 | Ph | VU0413255 | 23036271 | 90744391 | 3.75E-04 |
| 22 | 4-CH3 | VU0418000 | 46172903 | 96021116 | 2.58E-04 |
| 23 | 4-OCH3 | VU0418001 | 46172904 | 96021117 | 2.58E-04 |
| 24 | 4-i-Pr | VU0418002 | 46172905 | 96021118 | 4.45E-04 |
| 25 | 4-Br | VU0424412 | 46926617 | 99432375 | 7.87E-05 |
| 26 | 4-C(O)NH2 | VU0424413 | 46926619 | 99432376 | 5.00E-04 |
| 27 | 4-NH2 | VU0424414 | 46926621 | 99432377 | 6.05E-05 |
| 28 |
 | VU0424416 | 46926624 | 99432379 | 2.90E-05 |
| 29 | 4-CO2H | VU0424460 | 46926639 | 99432387 | Inactive |
| 30 | 4-N3 | VU0424461 | 46926641 | 99432388 | 5.80E-05 |
| 31 |
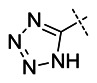 | VU0424462 | 46926643 | 99432389 | Inactive |
| 32 | 4-CH2NH2 | VU0424463 | 46926645 | 99432390 | 7.60E-06 |
| 33 | 4-CO2CH3 | VU0424464 | 46926647 | 99432391 | 8.60E-05 |
Table 3SAR of Scaffold 6 Diphenyl Carbinol
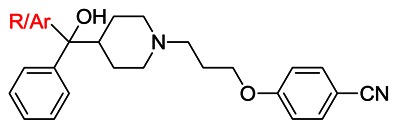 | |||||
|---|---|---|---|---|---|
| Entry | R/Ar group | VU ID | CID | SID | AVE IC50 (μM, n of 2) |
| 1 | H | VU0424409 | 46926611 | 99432372 | 2.34E-04 |
| 2 | n-Bu | VU0424410 | 46926613 | 99432373 | 1.53E-05 |
| 3 | cyclopropyl | VU0424411 | 46926615 | 99432374 | 1.12E-05 |
| 4 | -- (ketone) | VU0424454 | 46926627 | 99432381 | 2.75E-04 |
| 5 | CH3 | VU0424455 | 46926629 | 99432382 | 2.95E-04 |
| 6 | cyclopentyl | VU0424456 ML227 | 46926631 | 99432383 | 8.83E-07 |
| 7 | cyclohexyl | VU0424457 | 46926633 | 99432384 | 1.68E-06 |
| 8 | 4-CH3OPh | VU0424458 | 46926635 | 99432385 | Inactive |
| 9 | 4-FPh | VU0424459 | 46926637 | 99432386 | 7.75E-05 |
| 10 | R or S cyclohexyl (peak A) | VU0433663 | 49791887 | 103147637 | 5.70E-07 |
| 11 | R or S cyclohexyl (peak B) | VU0433664 | 49791888 | 103147638 | 1.13E-06 |
| 12 | RorScyclopentyl (peak A) | VU0433665 | 49791889 | 103147639 | 3.36E-07 |
| 13 | RorScyclopentyl (peak B) | VU0433666 | 49791890 | 103147640 | 4.13E-07 |
| 14 | i-Pr | VU0433667 | 49791891 | 103147641 | 4.05E-06 |
| 15 | cyclobutyl | VU0433794 | 49791893 | 103147642 | 4.00E-06 |
| 16 | CF3 | VU0433795 | 49791895 | 103147643 | 3.10E-05 |
3.5. Cellular Activity
ML227 was tested by MTT cell viability assay for its impact on cell proliferation in MLL leukemia cells harboring different translocations of MLL. Substantial growth inhibition was observed in three different MLL leukemia cells lines: MV4;11 (harboring MLL-AF4 fusion protein), ML-2 (with MLL-AF6 fusion protein) and KOPN-8 (with MLL-ENL fusion protein), with GI50 values at the range of 15–20 μM (Figure 12). In contrast, CID 46926611 (barcode R8Q), which shares the same core structure as ML227 but is missing the cyclopentyl group (SAR Table 3, entry 1), is a very weak inhibitor of the menin-MLL interaction (IC50 = 234 μM) and shows a very limited effect in MLL leukemia cells. These results demonstrate a strong correlation between the in vitro inhibition of menin-MLL interaction and inhibition of cell growth in MLL leukemia cells for this class of compounds. A broader collection of leukemia cell lines with and without MLL translocations will be tested to further assess specificity and toxicity of ML227.
Protocol for MTT assay
Cells were plated in 90 μL of culture medium in 96-well flat bottom microtiter plates (Fisher Scientific) at concentrations of 1 × 105/ml. Cells were treated with 0.25% sterile DMSO (Sigma) or serial dilutions of compounds from 20 mM stock solutions in DMSO (all final concentrations of 0.25% DMSO). Cells were incubated in a 5% CO2 incubator at 37 °C for 72h. A Vybrant MTT cell proliferation assay kit (Molecular Probes) was employed. Plates were read for absorbance at 570 nm using a PHERAstar BMG microplate reader. The experiments were performed in quadruplicate with mean and standard deviation calculated for each condition.
3.6. Profiling Assays
The enantiomers of the probe molecule (ML227) were tested at Ricerca’s (formerly MDS Pharma’s) Lead Profiling Screen (binding assay panel of 68 GPCRs, ion channels and transporters screened at 10 μM), and were found to have significant activity. Plans to address these activities are further discussed in section 4.3.
4. Discussion
This probe molecule (ML227) is a non-covalent inhibitor of the menin-MLL interaction. ML227 can be accessed synthetically in three-four steps in good overall yield. ML227 shows excellent inhibition in the HTRF assay and demonstrates moderate inhibition on cell proliferation in MLL leukemia cells harboring different translocations of MLL, including: MV4;11 (harboring MLL-AF4 fusion protein), ML-2 (with MLL-AF6 fusion protein) and KOPN-8 (with MLL-ENL fusion protein). ML227 is anticipated to lead to further interest in the development of small molecule inhibitors for the treatment of MLL associated leukemias.
4.1. Comparison to Existing Art and How the New Probe is an Improvement
Currently, there are no known publicly available small molecule PPI probes available which disrupt the menin-MLL interaction. The Assay Provider has patent applications (US 20090181917 and WO 2008070303) which describe cell-permeable peptides that disrupt the menin-MLL interaction. Recently (March 10, 2011) a patent application, WO 2011029054, from the Assay Provider’s group (former institute, University of Virginia) published an International PCT describing independently identified compounds structurally related to ML HTS Scaffold 4. (21) A handful of compounds are described as being sub-micromolar in this application. At this time and based on the current data available it is not clear if ML227 has advantages over the subject matter claimed. Ongoing collaborative efforts and discussions are underway to decide how to proceed with a potential narrow patent application and publication strategy around ML227.
4.2. Mechanism of Action Studies
As described in the significance section, the menin interaction with MLL is critical for the oncogenic activity of MLL fusions, validating the menin-MLL interaction as a potential target for molecular therapy. Inhibition of the association of menin with both MLL and MLL fusions by small molecule inhibitors is hypothesized to be useful as a new therapeutic approach to treat MLL-associated leukemias. STD NMR studies were used to validate a competitive non-covalent interaction with menin and lead compound MLS001171971 (Scaffold 6), suggesting a physical binding interaction with the menin protein. Efforts are ongoing to conduct further STD NMR studies using ML227 itself in order to gain a better understanding of the nature of its interaction with menin. Furthermore, the strong correlation between the biochemical and cellular inhibition of cell growth in MLL leukemia cells for ML227 is strongly supportive of this mechanism.
4.3. Planned Future Studies
Structurally, elements of ML227 and its HTS lead MLS001171971 share feature related to a well known class of antihistamines and other related monoamine GPCR orthosteric ligands. These compounds, including terfenadine (Seldane) and the second generation antihistamine fexofenadine (Allegra) for example (Figure 13), often have a rich poly-pharmacology but principally there efficacy in treating allergy symptoms is thought to be due to their antagonism of the H1 receptor.
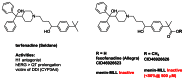
Figure 13
ML227 pharmacophore related H1 antagonists.
Early on, we obtained and tested the active metabolite and methyl ester of terfenadine- CID 46926623 (fexofenadine) and CID 46926626 (Figure 13). Neither of these compounds were active as a menin-MLL PPI disruptor. In light of the ancillary profile associated with the MLL227 enantiomers, including H1 antagonism, we aim to pursue known strategies to mitigate the monoaminergic receptor and ion channel activity found in ML227 (22 and references therein.) A major physiochemical feature of ML227 which gives rise to these activities can be attributed to its high lipophilicity (cLogP > 5.0) in conjunction with the highly basic piperidine nitrogen (pKa ~ 8.6). Shown in Figure 14 is a summary of recent compounds in hand awaiting testing along with selected targets which incorporate structural features designed to reduce cLogP and/or reduce pKa of the central piperidine nucleus. In general, we have not established the necessity of the piperidine nitrogen and therefore there are a number of possible piperidine replacements to examine. In particular, the hydroxyl cyclohexane derivative highlighted should be particularly informative. Incorporation of the linker alcohol found in the original lead CID 4453701 in combination with the potency enhancing cyclopentyl group appears to be an obvious hybrid target. In addition, the 1,2,3-triazole replacement (CID 46926624, SAR Table 2) represents only a ~3× loss relative to cyano group and will be pursued in the same way along with various 1,2,4-heterocycles which thus far have not been examined.
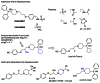
Figure 14
Recent analogs and selected future targets.
5. References
- 1.
- Sorensen PH, Chen CS, Smith FO, Arthur DC, Domer PH, Bernstein ID, Korsmeyer SJ, Hammond GD, Kersey JH. J Clin Invest. 1994;93:429–437. [PMC free article: PMC293805] [PubMed: 8282816]
- 2.
- Cox MC, Panetta P, Lo-Coco F, Del Poeta G, Venditti A, Maurillo L, Del Principe MI, Mauriello A, Anemona L, Bruno A, Mazzone C, Palombo P, Amadori S. Am J Clin Pathol. 2004;122:298–306. [PubMed: 15323147]
- 3.
- Eguchi M, Eguchi-Ishimae M, Greaves M. Int J Hematol. 2003;78:390–401. [PubMed: 14704031]
- 4.
- Greaves MF. Leukemia. 1996;10:372–377. [PubMed: 8637251]
- 5.
- Slany RK. Hematol Oncol. 2005;23:1–9. [PubMed: 16118769]
- 6.
- Tkachuk DC, Kohler S, Cleary ML. Cell. 1992;71:691–700. [PubMed: 1423624]
- 7.
- Sedkov Y, Tillib S, Mizrokhi L, Mazo A. Development. 1994;120:1907–1917. [PubMed: 7924996]
- 8.
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Nature. 1995;378:505–508. [PubMed: 7477409]
- 9.
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Proc Natl Acad Sci U S A. 2005;102:8603–8608. [PMC free article: PMC1150839] [PubMed: 15941828]
- 10.
- Hess JL. Crit Rev Eukaryot Gene Expr. 2004;14:235–254. [PubMed: 15663355]
- 11.
- Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, Hess JL, Slany RK. Mol Cell Biol. 2004;24:617–628. [PMC free article: PMC343796] [PubMed: 14701735]
- 12.
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Mol Cell. 2004;13:587–597. [PubMed: 14992727]
- 13.
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. Cell. 2005;123:207–218. [PubMed: 16239140]
- 14.
- Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Cancer Res. 2007;67:7275–7283. [PMC free article: PMC7566887] [PubMed: 17671196]
- 15.
- Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. Proc Natl Acad Sci U S A. 2006;103:1018–1023. [PMC free article: PMC1326489] [PubMed: 16415155]
- 16.
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Mol Cell Biol. 2004;24:5639–5649. [PMC free article: PMC480881] [PubMed: 15199122]
- 17.
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Science. 1997;276:404–407. [PubMed: 9103196]
- 18.
- Marx SJ. Nat Rev Cancer. 2005;5:367–375. [PubMed: 15864278]
- 19.
- Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. Cancer Cell. 2010;17:148–159. [PMC free article: PMC2830208] [PubMed: 20159607]
- 20.
- Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. John Wiley & Sons, Inc.; 1994. pp. 606–613.
- 21.
- Ekins S, Crumb WJ, Sarazan RD, Wikel JH, Wrighton SA. J Pharm Exp Ther. 2002;301:427–434. [PubMed: 11961040]
- 22.
- Hess J, Grembecka J, Cierpicki T. Compositions and Methods for Treatment of Leukemia. WO 2011029054. PCT Int Appl. 2011
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Progress towards small molecule inhibitors of the Menin-Mixed Lineage Leukemia (MLL) interaction with in vivo utility.[Probe Reports from the NIH Mol...]Review Progress towards small molecule inhibitors of the Menin-Mixed Lineage Leukemia (MLL) interaction with in vivo utility.Senter T, Gogliotti R, Han C, Locuson CW II, Morrison R, Daniels JS, Cierpicki T, Grembecka J, Lindsley CW, Stauffer SR. Probe Reports from the NIH Molecular Libraries Program. 2010
- Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein-Protein Interaction.[J Med Chem. 2019]Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein-Protein Interaction.Aguilar A, Zheng K, Xu T, Xu S, Huang L, Fernandez-Salas E, Liu L, Bernard D, Harvey KP, Foster C, et al. J Med Chem. 2019 Jul 11; 62(13):6015-6034. Epub 2019 Jun 22.
- Identification of novel small-molecule inhibitors targeting menin-MLL interaction, repurposing the antidiarrheal loperamide.[Org Biomol Chem. 2016]Identification of novel small-molecule inhibitors targeting menin-MLL interaction, repurposing the antidiarrheal loperamide.Yue L, Du J, Ye F, Chen Z, Li L, Lian F, Zhang B, Zhang Y, Jiang H, Chen K, et al. Org Biomol Chem. 2016 Sep 28; 14(36):8503-19. Epub 2016 Aug 19.
- Progress towards small molecule menin-mixed lineage leukemia (MLL) interaction inhibitors with in vivo utility.[Bioorg Med Chem Lett. 2015]Progress towards small molecule menin-mixed lineage leukemia (MLL) interaction inhibitors with in vivo utility.Senter T, Gogliotti RD, Han C, Locuson CW, Morrison R, Daniels JS, Cierpicki T, Grembecka J, Lindsley CW, Stauffer SR. Bioorg Med Chem Lett. 2015 Jul 1; 25(13):2720-5. Epub 2015 Apr 25.
- Review Menin-MLL protein-protein interaction inhibitors: a patent review (2014-2021).[Expert Opin Ther Pat. 2022]Review Menin-MLL protein-protein interaction inhibitors: a patent review (2014-2021).Bai H, Zhang SQ, Lei H, Wang F, Ma M, Xin M. Expert Opin Ther Pat. 2022 May; 32(5):507-522. Epub 2022 Mar 11.
- Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction - Probe Reports...Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...