Abstract
Mass Spectrometry based screening has allowed researcher’s to employ native substrates for screening, screen previously intractable targets, and eliminate the need to use coupled reactions. Recently, many instrumentation advances have been made to increase throughput, thus allowing routine access to this technology. This chapter details the different instrumental set-ups which will enable mass spectrometry based screening, and issues and solutions likely to be encountered during assay development.
Introduction
The use of LC-MS/MS as an assay platform for hit generation has enabled the development of assay methodologies that were previously difficult or impossible using conventional means of detection (2-4). These instruments are able to identify and quantify low levels of analytes in complex matrices (5). Their ability to detect unmodified natural substrates and products is also an advantage as modified substrates may impact the quality of hits (6, 7). Additionally, the near universality of detection gives LC-MS/MS a wide range of applications. Historically, LC-MS/MS detection techniques were too slow for screening applications. However, with the clever use of valves and pumps, several researchers are beginning to exploit LC-MS/MS and have implemented this strategy for routine screening (8-10). Liquid chromatography techniques encompass no chromatography (flow injection), to isocratic, and to gradient methods using short columns, and to staggered injections on multiple columns. Early versions of LC-MS/MS screening utilized Gilson’s 8-probe liquid handler and staggered injection systems (8, 9). A few years ago, a company employing a unique autosampler, (RapidFire™, Agilent) was spun out of Pfizer with LC-MS/MS screening as their mainstay (10). In the past few years, other manufacturers have offered autosamplers capable of the high sampling rates required for this technique. This manual will focus on four systems which are proven to deliver the through-put necessary for hit generation for a 100,000-200,000 compound screen in a few weeks. Also, the technical details ranging from assay buffer components to data processing will discussed.
Assay Components
Ideally, only buffers and detergents which are compatible with the mass spectrometer should be used in the bio-chemical assay mixture. However, enzyme systems commonly require the presence of non-volatile buffers, proteins, and/or detergents for optimal activity. The levels to which non-volatile components can be tolerated in the bio-chemical system is directly related to the chromatographic system employed (). For example, the flow injection mode is most sensitive to ion suppression and requires the most sample clean-up or only mass spectrometry friendly components. Whereas chromatographic separation methods can tolerate less stringent sample clean up and less mass spectrometry friendly components can be employed.
Volatile HPLC Modifiers used in LC-MS/MS
The buffers of choice for the biochemical assay are ammonium acetate and ammonium bicarbonate as these buffers are compatible with the mass spectrometer due to their volatility. If these buffers are not compatible with the enzyme assay, then conventional buffers can be used. However, for flow injection significant sample cleanup would need to occur. Detergents represent the most troublesome assay components as these are normally retained on reverse phase resins. Typically, replacing or avoiding SDS and polymeric detergents such as TWEEN, Triton, and NP50 is the best course of action. Polymeric surfactants are mixtures of several molecules of varying polarities and masses, which interfere by eluting in a broad time window and flooding the system with a large number of ions that will greatly suppress the desired signals or cause “cross talk”(11). If these cannot be replaced with the more mass spec friendly detergents, then these should be used at the lowest effective concentration. When detergents must be used, single molecule forms are best e.g. CHAPS, octyl β-D-glucopyranoside or dodecyl β-D-glucopyranoside. Then, only one component needs to be separated from the analytes to enable detection.
Mass Spectrometers
The choice of mass spectrometer will have significant impact on the ability to perform MS based screening. A triple quadrupole is the preferred instrument because it delivers the best combination of sensitivity, selectivity, scan speed, and dynamic range. A triple quadrupole instrument is usually run in multiple reaction monitoring (MRM) mode. MRM works by selecting the ion of interest in the first quadrupole, fragmenting the ion of interest in the second quadrupole, and finally selecting one of the fragments in the third quadrupole for detection by the detector. Typically, one unique product ion is selected for each parent, but more than one product ion can be selected as a confirmatory ion. Also, quadrupole-time-of-flight or orbit trap instruments are being used for screening. Quadrupole-time-of-flight or orbit trap instruments provide the ability to obtain accurate mass measurements of the ions of interest. Due to this capability a narrow mass range window can be used to obtain specificity rather than fragmenting the molecule as in a triple quadrupole instrument. This can be a significant advantage when dealing with small molecules which have poor fragmentation efficiency or do not fragment to produce a dominant ion (12). The mass range, resolution, and speed of the quadrupole-time-of-flight or orbit trap enable the use of small proteins as substrates that may generate multiple enzymatic products. It is not necessary to have standards for the products as the entire mass range is captured in the TOF scan, enabling all the products to be measured simultaneously. For example, a small protein which undergoes multiple phosphorylations could be analyzed directly to determine if inhibitors give different phosphorylation patterns. The drawbacks to the use of Q-Tof instruments for screening are the large data files produced and the limited data processing software available. However, instrument companies are activity working to remove these barriers.
The short LC columns or flow-injection used in mass spectrometry based screening places additional demands and restraints on the mass spectrometry system. The reduced chromatography used in these rapid assays typically does not separate substrate and the product, therefore; the use of unique MRM transitions or exact masses for identification and quantitation are needed. Generally, no problems are seen if the compounds are not isobaric as unique molecular ions are observed. Occasionally, cross-talk between analytes may occur if the collision cell of the MS is not sufficiently cleared between analyte transitions (seen only in older instruments), or if an analyte converts to the other analyte in the source. If analytes have the same molecular ion and the fragment selected for analyte A also appears as a fragment for analyte B, then a pure sample of A will produce a peak in B and vice versa. If A and B are separated in time, this is not a problem. However, analytes are normally not separated in these rapid assays and unique fragments are then required. This may necessitate the use of lower sensitivity fragments to assure uniqueness. Typically, sensitivity is not an issue for these screening assays. As an illustration, a major fragment at 271 is seen in the analysis of several prostaglandins that would be optimal for sensitivity; if the analyte did not co-elute with other components also producing this fragment. Selection of other less abundant fragments allows unique identification of specific prostaglandin analytes. The isomeric prostaglandins PGE2 and PGD2 may be identified by their fragmentation pattern (13). Also the 5-, 12-, and 15-HETEs are good examples of isobaric molecules which can be identified and quantitated by their fragmentation patterns even through signal intensity is lost compared to the most abundant fragment (14).
To deal with the inherently variable process of generating and measuring ions in a mass spectrometer, internal standards are used. The best internal standards are isotopically labeled analogues with a mass shift of +4 or greater. If an isotopically labeled product is not available, then a structural analog should be used. The internal standard should be added as soon as possible, (normally when the reaction is quenched) and its concentration should be half way between the maximum and minimum concentration of the product. The internal standard normalizes the analyte response to a known amount of material that has undergone identical sample handling. Therefore, once the internal standard is added, the ratio of analyte to internal standard is fixed regardless of decomposition, sample handling differences, and variances inherent in the mass spectrometry process. If the substrate and product for a reaction are close in structure and the calibration curves are parallel then the substrate can be used as in a ratio with the product. If the substrate is completely different than the product or the product and substrate calibration curves are not parallel, then an internal standard should be used.
The high flow rates used to obtain the through-put necessary to enable an MTS campaign place increase desolvation demand on the mass spectrometer. The source temperature is usually set to the highest allowed value and the source gas flow rates are set very close to the maximum allowed. Also, the high volume of samples injected on the mass spectrometer is greater than the typical sample work load. These special circumstances necessitate a more frequent cleaning schedule compared to that typically recommended by the instrument vendor. When running 100 or more 384-well plates a week, the instrument should be cleaned every two weeks or earlier depending upon the signal degradation. This can be monitored by plotting the response of the control wells over time. When the area counts decrease twenty-five percent or more then cleaning should occur.
For typical bioanalytical assays, linearity of triple quadrupole instrument response to analyte concentration may be established over >4 orders of magnitude. For screening assays, only 2-3 orders of magnitude are needed as activity of the reaction is measured from 0-100% inhibition. It is important to monitor the analytes in the linear range of the instrument response because calculation of percent inhibition assumes a linear function. Also, the signal to noise ratio for the product should be sufficient to measure 99% inhibition of the enzyme. The response of the non-inhibited wells should therefore be at least 100-fold signal to noise.
The mass spectrometer can ionize molecules using four basic modes of ionization electrospray positive, electrospray negative, atmospheric pressure chemical ionization positive and atmospheric pressure chemical ionization negative. While any ionization mode may be used, typically, electrospray ionization is used; as most compounds will have a fixed or inducible charge or be able to form adducts with an appropriate modifier. The topic of electrospray ionization is beyond the scope of this article. Here are a number of reviews and books on this topic (15-18)
Sample Preparation
Normally, some clean-up of screening samples is required prior to chromatography (). At the very least, a stop reagent that halts the reaction and engenders stability to the final mixture is required, as the samples may sit in the autosampler or freezer for an extended time. The stop reagent may consist of a low or high pH buffer, organic solvent, quenching reagent, derivatizing reagent, etc; similar to normal screening methods. Again, volatile reagents should be used to avoid contamination of the MS system. The internal standard(s) is normally added in the stop reagent (see MS parameters section).
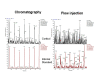
Comparison between ballistic chromatography and flow injection for 10 ng/mL of cortisol. Intensity of internal standard for flow injection was 2.6e4 compared to 5.4e5 for the ballistic chromatography setup.
The most common stop reagents are acetonitrile or methanol with or without modifier, and water with formic or trifluoroacetic acid. Acetonitrile (ACN) or methanol ratios of 1:1 to 1:4 sample to solvent are generally used. The high levels of ACN or methanol will cause the proteins in the sample to precipitate, and require the plates to be centrifuged or filtered prior to analysis. If the biochemical system cannot be stopped with ACN or methanol due to chromatographic issues, then water with formic or trifluroacetic acid at 1-10% (v/v) should be tried. This type of sample preparation leaves the salts and buffers in the sample. Therefore, the system must include a divert valve or “dump valve” between the column and the MS to remove the salts to waste and preserve the cleanliness of the MS. Alternatively, the analytes may be captured from the assay mix by liquid/liquid or solid phase extraction, followed by drying and reconstitution or dilution with water. Analysis of the subsequent samples often does not require a “dump valve” system as these methods result in cleaner samples with salts, proteins, and detergents typically removed. Liquid/liquid extraction methods may be automated using robotics, but require easily achieved modifications to the normal robotics and use of solvent resistant plates. Venting of the robot to remove organic solvent(s) vapors is necessary for safety (; see instrument set-up section). Also, organic solvents used in liquid/liquid extraction are not tolerated by the plates normally used for the assays . Of the common polymers used, only polypropylene is resistant to some of these solvents (see manufacturer’s specifications). However, Teflon-like plates are available in 96 and 384 formats. These Teflon-like plates are typically used for combinatorial chemistry applications, and are more expensive than the plates normally employed in screening. However, in many cases, they may be washed and re-used. Also, reagent reservoirs containing solvents on the robot deck must also be made of solvent-resistant materials (e.g. metal, glass, Teflon). As pipette tips are constructed from polypropylene, they tend to be resistant to solvents, especially as they are only in contact with the solvent for a short time. However, compatibility should be checked prior to use. Although 96 and 384-well plate based SPE phases are available, these tend to be cost prohibitive when considering the number of samples to be processed in a typical screen.
Schematic for Modified BioMek FX for Organic Solvents
Derivatization of the analytes can make them amenable to extraction or detection. This is beneficial when the analytes are difficult to chromatograph due to sticking to surfaces, broadening of peaks, or poor retention. However, this does add further complexity to the screen and may affect the assay statistics if the reactions are not optimized or complete. If derivatization is required for sensitivity or to overcome some other property, both substrate and product should react to the same level of completion or a stable-labeled internal standard will be necessary.
Chromatography
In the most common situation, chromatography is required to avoid ion suppression from the sample matrix (). The goal is to keep the analysis time as short as possible while providing acceptable separation of the analytes from the suppressing contaminants. A chart of sample analysis cycle time and daily throughput is shown in . In the realm of analysis times <20 seconds/sample, assay screens of 50-100K samples become feasible. However, even an increase of a few seconds/sample will raise the total screen time significantly. This restricts the use of chromatography to ballistic or snap gradients – essentially “on-line SPE” with little or no separation of the analytes from each other, requiring unique MRM transition channels for each analyte or unique parent masses for accurate mass instruments. In these methods, the sample is loaded with a retaining mobile phase while unretained components are diverted to waste (via a dump valve), followed by a very rapid mobile phase switch to elute the analytes to the MS.
The two most used types of chromatography for medium through-put screening are reverse-phase and HILIC. Reverse-phase chromatography uses a hydrophobic stationary phase, usually a C18 alkyl chain bonded to a silica particle. The C18 alkyl chain can be replaced by a C4, C8, phenyl, or cyano, but the same methodology applies. The initial mobile phase is water with or without some amount of organic, plus a modifier. Typical modifiers and their concentrations are located in the table (). The elution solvent is then acetonitrile or methanol with the same or possibly different modifiers. The analytes are retained on the alkyl phase due to its hydrophobic nature and then elute with the higher percentage of organic solvent. This type of chromatography is excellent for the majority of analytes which will be screened by mass spectrometry. Reverse phase chromatography is also versatile due to the ability to change modifiers which adjust the retention of the analytes. Two of the most significant advances with this type of chromatography are the increase in pH range and the ability to use 100 percent aqueous solvent systems. The bonded phase is typically only stable to pH 7; however, with the introduction of hybrid particles, pH stability up to twelve is possible. This allows for the use of high pH mobile phases to retain molecules that are not well behaved at lower pH. Also, increase in pH can dramatically improve peak shape and can sometimes dramatically effect sensitivity in the mass spectrometer. Using 100% aqueous phases may cause phase collapse for older resins, resulting in non-reproducible retention times. However, resins are currently manufactured which are compatible with 100% aqueous phases. For a more in-depth discussion of reverse-phase chromatography, please refer to the following references (19-26).
HILIC (Hydrophilic Interaction Chromatography) is very good for retention of polar analytes. This type of partition chromatography can use the same solvents as reverse-phase chromatography but in the reverse order. The compounds are loaded in high organic (typically 95% acetonitrile) and eluted by increasing the water content. The samples are also typically in a high percentage of organic solvent, which couples well with stopping the reaction with acetonitrile and then direct analysis of the samples. The stationary phases for HILIC are quite different from reverse-phase chromatography. Typically HILIC stationary phases are bare silica, diol, amino, amide or specialty stationary phases which have dual functional groups. Unlike reverse-phase columns where exchanging one manufacturer’s C18 column for another may provide minimum changes in peak shape or retention, changing from one HILIC packing material to another may produce significant changes in retention, peak shape, and carry-over. For this reason, typically 5 or 6 different HILIC stationary phases are tested using the same mobile phase composition. For a more in depth discussions on HILIC chromatography please refer to these reviews (23, 25, 27, 28).
Instrument Set Up
In this section, several arrangements of instruments are described. Although other arrangements are possible, these have proven robust in our hands.
Modified Gilson 8-probe
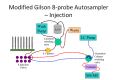
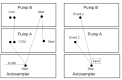
For extracted or generally clean samples, an isocratic system, developed employing a Gilson 8-probe that uses a mobile phase and column allowing for slight separation of the void volume and analytes (8). The setup is pictured in Figures and . The Gilson 8-probe was modified by addition of a 10-port column-switching valve that is used to route pump flow to each of the eight sample injection valves in sequence, each sample injection valve linked directly to the corresponding positions on the switching valve. The two remaining positions on the switching valve (9 & 10) are used to by-pass the Gilson, routing flow directly to the column. During operation, the eight samples are loaded to the corresponding sample injection valve loops (load position) with the switching valve in the by-pass position (#10). Then, all sample valves are switched to the inject position. Because there is no flow through the sample valves, the samples are locked in the loops. Each sample valve is then selected in turn by rotating the switching valve through positions #1-8 using a time program of contact closures from the Agilent HPLC pump. Position #9 is passed over to wait at #10 for the next eight samples. As each sample injection valve is selected, the isocratic flow (2 ml/min) sweeps the sample from its loop to the column, where it is separated, and on to the MS, where it is detected. Each sample valve is selected for 6 seconds. The analytes are partially retained on the column, separating slightly from the void volume. The pressure spike observed on switching the 10-port valve causes a blip in the data trace from the MS and timing of its occurrence is adjusted away from the appearance of the peaks to enable facile integration.
Schematic of Modified Gilson 8-probe Autosampler in the injection position. All peek lines have been replaced metal.
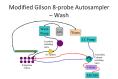
Schematic of Modified Gilson 8-probe Autosampler in Wash position. All peek tubing has been replaced with steel.
During the rotation of the switching valve, the Gilson’s 8-probes and injection ports are washed using a modified washing system. The needles are washed at the wash station, both inside and out, using the syringe working solvent in the normal manner. The injection ports are washed using a modified washing system incorporating a HPLC pump, a two-position switching valve, and an 8-way splitter. Flow of wash solution from the pump (16 ml/min) proceeds through the two-position switching valve that directs flow to the pump solvent reservoir or the injection ports, thus recycling solvent when not in use. When the injection ports are selected, flow proceeds through the 8-way splitter creating a flow of 2 ml/min to the exit/waste ports of the sample valves and up through the needle entrance of the ports, back-flushing the ports. The overflow from the ports is collected in the drainage moat surrounding the ports and exits via a drain tube to a waste bottle that is under constant negative pressure using an exhaust trunk. This method reduces carryover to essentially zero and also clears any plugging of the injection ports.
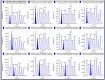
For high-throughput operation, the Agilent HPLC pump, degasser, and column compartment, and the AB Sciex API-4000 are controlled by the Sciex Analyst software. To facilitate the rapid analysis, data was acquired into a single data file for each iteration of the Gilson 8-probe, yielding 8 samples per file (). The movement of the column switching valve used to select the individual sample injection valves on the Gilson 8-probe was controlled by the inject start contact closure from the 8-probe and a time program of contact closures from the Agilent HPLC pump. The washing pump was set up to run continuously, with its switching valve controlled via the AB Sciex software. The Gilson 8-probe was run in stand-alone mode using Trilution software, but with a Ready-Out contact closure signal required from the LC-MS/MS. The 8-probe also sent an inject start contact closure to the MS to start the acquisition. Thus, if the autosampler failed, the MS would not start. Also, if the LC-MS/MS failed, the autosampler would not proceed. This dual “handshake” saves samples in the event of equipment failure. To prevent failures due to leaks in the plumbing that are missed by the leak detection resistors on the Agilent equipment, the low pressure setting on the pump is set to 34 bar. Also, as PEEK tubing will fail after prolonged exposure to ACN, all lines up to the column were plumbed with stainless steel. Data handling utilized a method written in the vendor software with analytes 1-8 representing samples 1-8 of each iteration of the 8-probe. After integration, the results were transferred to Excel and transposed with a template to 384-well format.
Example of data obtained with Gilson 8-probe. Gilson 8-probe was connected to AB SCIEX 4000 and the data was processed using Analyst®.
Off-the-shelf MTS system
For analyses requiring a ballistic gradient, the Shimadzu Prominence system offers a practical alternative. A typical set up utilizes a 10-port two position valve to select pumps and route flow to either waste or the MS. During equilibration and sample injection, flow proceeds from the loading LC pump to the autosampler, to the 10-port valve, through the column, and exiting to waste. Flow from the eluting LC pump proceeds through the same 10-port valve to the MS. After the sample is injected, the analytes are retained on the column while unretained materials are sent to waste. The valve switch moves the column from the loading pump flow to the eluting pump stream and sweeps the analytes into the MS. After the analytes have eluted, the valve is switched again and the column re-equilibrated with the loading pump flow.
illustrates the layout of the pumps, autosampler, 10 port valve and mass spectrometer. This arrangement provides for instantaneous delivery of the elution and re-equilibration solvents to the column through the 10 port valve. Also, solvent B is continuously pumped to the MS, either bypassing the column or through the column.
Schematic of MS-MTS system using the 20 AC Shimadzu auto-sampler, 20 AD Shimadzu pumps, 10 port value, and a mass spectrometer.
depicts the wiring diagram which starts both the pumps and the mass spectrometer simultaneously. With each injection, the autosampler sends a start signal to the pumps to begin the pump’s time programs and to the mass spectrometer. However, the mass spectrometer ignores any further start signals from the autosampler until the time programmed into the mass spectrometer method has elapsed. The time program for pump A controls the switching valve and the pump’s shut off time. The time program for pump B controls its shut off time. The flow rates for pump A and B are 2.5 mL min-1 until 2.9 minutes, with a drop to 0 mL min-1 at 3 min. With this set-up the pumps will stop flowing if they do not receive a start signal before 3.0 minutes. Therefore, after the 12th plate has run or if an error occurs, the pumps will shut off, allowing for unattended operation of the system.
Schematic of wiring. A) Wiring setup for starting the pumps and auto-sampler simultaneously. B) Wiring setup for sending a stop signal to the auto-sampler if the pumps’ pressure fall below minimum pressure setting, or detects a leak.
depicts the wiring diagram enabling a pump error to stop the autosampler while running in local mode. After the wiring is connected, the External Signal Functions (EXT-S) parameter in each pump is set to 2. Without this configuration, the autosampler would continue to inject samples even though the pumps are not working. There is no need the change the wiring connections and the EXT-S settings when switching between MTS mode and normal bio-analytical mode.
Commercially-built System for MTS
A commercially available instrument was built to perform rapid ballistic gradients with a computerized interface to facilitate automated mass spectrometry based screening. Therefore, this instrument has significant advantages in ease of use, speed, and robustness over the previous two platforms. The instrument consists of three isocratic pumps and one peristaltic pump, four valves, a vacuum system for sample loading, column holder, refractive index detector, and a plate changer. Another vital component is the software to integrate the data which is further discussed in the data handling section. Unlike most sample inlet systems, this commercially built instrument uses vacuum to remove sample from the sample plate. The sample is pulled through the tubing, through the injector loop, and to an optical window. A refractive index change is detected when the sample liquid appears, which then triggers the injection valve to switch and load the column with the sample. If the sample is not loaded by the time specified in the method, the valve will still inject and that well will be missed. If a specified number of missed injections occur, the system will shut down. The three isocratic pumps work in concert with the values to provide low carryover and rapid sample clean up. Pump 1 is used to load the compound onto the column, wash the sample, and re-equilibrate the system. Pump 2 is used to wash the sample flow path. The solvents from pump 2 do not interact with the sample. This allows for the possibility of using a very aggressive wash in pump 2 if needed to reduce carryover; however, this solvent is usually the same as the elution solvent. Pump 3 elutes the sample from the column, with flow either to the mass spectrometer or through the column to the mass spectrometer. When the system switched from load to elute, pump 2 is then flushing the system while pump 3 is eluting the sample. The column compartment on this instrument can hold up to 6 columns. When performing method development these can range from cyano to C18 phase. In production mode these are the same stationary phase. If during a run the backpressure is above the limit specified and there is another column of the same packing material available, then the instrument will automatically change to the new column and resume the analysis. The standard packing materials supplied in their catalog are cyano, C4, C8, C18, silica, HILIC and Hypercarb columns. Custom packed columns can be obtained; however, by modifying the plumbing the system can be made to accept a multitude of columns. This is especially useful when trying to decide on the best column or if a high pH compatible stationary phase either for reverse-phase or HILIC is necessary. The one drawback to modifying the plumbing is the loss of the automated column switching capability. But the ability to use more column types is a worthwhile trade off. This system is best illustrated by the Agilent Rapidfire™ (2, 4, 29-31).
Staggered Injection System
The staggered injection system is the only true HPLC system of the four discussed. These systems are typically composed of four HPLC pumps, two or four auto-samplers, four columns, a column switcher, and advanced software to integrate and control these components (32-34). The injections are staggered in time so that the compounds are eluting at different times into the mass spectrometer. These systems allow true gradient elution instead of only a ballistic gradient. Therefore, this type of system may be able to successfully overcome more complex matrixes than the systems previously described. The price paid for this increased chromatographic resolving power is speed. The fastest this system can inject is about one sample per minute; therefore, the best through-put this system can achieve is 4-5 samples per minute. The biggest shortcoming to this system is all the parts that have to keep running to achieve this through-put. Recent versions of software controlling this system will compensate for loss of one channel by redistributing the samples across the other three channels. This system (like the Shimadzu system previously discussed) can be used to perform other functions besides MTS screening making it a versatile option for any lab thinking about screening on infrequent basis. This system is best illustrated by the Aria System from Thermofisher.
Data Handling
In years past, processing MTS data was a challenge using vendor software; however, this is now not the case. The most complete solution is provided using the Rapidfire® because that system has its own data integration software capable of integrating data from Thermo Fischer Scientific, AB Sciex and Agilent mass spectrometers. Using the Agilent platform integration software it is quite easy to process data from samples contained in 10 to 20 plates collected in a single file. After straight-forward data integration, the software can display the data in either plate format or column format. The software will do an excellent job processing data in screening mode; however, if calibration curves are needed, then the software has limitations depending upon the MS instrument used. If the Rapidfire® is connected to an Agilent mass spectrometer, then making in-plate calibration curves will be straight-forward as the system is fully integrated into the Agilent Mass Hunter software. If the Rapidfire® is connected to a Thermo Fischer Scientific or AB Sciex mass spectrometer, obtaining a calibration curve requires data to be imported into a program like Excel or JMP for further processing or will require using the mass spectrometry vendor’s software to process the data.
All three brands major brands of mass spectrometers (Agilent, ThermoFisher Scientific and AB Sciex) may be used to obtain data from the Gilson 8-probe or the Shimadzu autosampler options described. Using the Gilson allows the normal vendor quantitation software to be used for data integration of the eight peaks per data file, with further processing in Excel or similar programs. The Shimadzu set up requires use of vendor specific software to process the data as the entire plate data is contained in a single file. For the Agilent, this will be done using their Mass Hunter software. For the ThermoFisher Scientific instrument, data processing is performed using GMSU/QuickCalcCalculation, a separate software package from their standard package. For AB Sciex, data processing is performed using their Multiquant™ software package, also purchased separately. As opposed to the Rapidfire® integration software, these vendor solutions work better if each 384/96 well plate is contained in a single file. These software packages also work better if an internal standard (or the substrate which can act as the internal standard) is used as an injection marker for processing the data. After the data is processed and exported to a spread sheet, percent conversion or area ratio can be calculated. Mass spectrometry data running in MTS mode is inherently more variable when only the product of the reaction is considered; however, because the product of the reaction is made from the substrate, a percent conversion calculation may be used to normalize the data. This should only be done if the instrument response slopes for the substrate and product standard curves are parallel. If the lines are not parallel an internal standard must be used. The calculation for percent conversion is:
% Conversion = Product/(Product + Substrate)
If the biochemical system allows for percent conversion to be used, this will result in the least amount of assay variance as it negates the variance caused by the robotics systems used to add substrate to the assay plates. Addition of internal standard will only account for the variance occurring after its addition at the stoppage of the reaction.
Other Notes
Some time is generally required for the screening lab personnel to become accepting of this new technique and its ability to pinpoint problem areas in assay methods and practice. In this process, robust experiments with adequate controls are needed that allow the data to speak for itself without equivocation. MRM transitions may be included that monitor each component of the reaction, allowing any errors or trends to be seen and facilitating rapid assay development. This will quickly highlight the new LC-MS/MS techniques as rapid assay development tools.
Collaborative efforts initiated with analytical departments containing the equipment and assay personnel are recommended as the purchase of the LC-MS/MS equipment requires significant capital expenditure, and its operation requires the incorporation of trained, experienced staff with the screening lab personnel.
Example Initiation of a New Screen
The following is a typical example of what occurs when developing a new MTS assay:
- 1.
Order product or products of the reaction and obtain substrate or substrates.
- 2.
Infuse the substrate/s and product/s into the triple quadrupole instrument to obtain the MRM transitions. Ascertain from infusion if molecule will ionize in positive mode only or negative mode only or both. Usually three MRM channels are obtained per analyte and tested.
- 3.
From the structure or from literature discern if reverse-phase or HILIC chromatography will retain and elute the compounds.
- 4.
Using a traditional HPLC system run a quick gradient from 100 percent water to 95 percent organic in reverse-phase mode or 95 percent organic to 10 percent organic in HILIC mode. Determine if peak is eluted from column with good peak shape and low carry-over. Determine minimum amount of organic tolerated for reverse-phase ie. 95/5, 90/10, 80/20 water/organic.
- 5.
Inject the substrate at 1 µM concentration to determine if cross talk into the product channel occurs. If there is less than 0.5% cross talk then proceed to next step. If significant cross talk is observed determine the reason.
- a.
Substrate is not pure and contains some product. This analysis needs chromatographic resolution between the product and substrate.
- b.
The MRM channel chosen is not specific enough. Re-infuse the analytes and obtain more MRM channels to test.
- 6.
Run a calibration curve in the assumed range of the reaction. We usually prepare our calibration curves by serial dilutions either 1:2 or 1:3 dilutions in water for reverse-phase and 1:3 water/ACN for HILIC. This will generate an ideal state/sensitivity.
- 7.
Determine if the reaction can be stopped with organic and at what ratio or does the reaction have to be stopped with water which will usually contain 10% formic acid in a 1:1 ratio.
- 8.
Rerun calibration curve in buffer system stopping with conditions developed in step 7.
- 9.
Now decrease the chromatographic run time by doing a step gradient i.e. hold for 6 seconds to wash and then elute at 90/10 organic/water for reverse-phase and 10/90 for HILIC.
- 10.
Rerun calibration curve with substrate and product to determine new limit of quantitation, linearity, and parallelism of substrate and product.
- 11.
If Substrate and product response are parallel, use percent conversion for screen. If slopes are not parallel, then obtain an analog IS or stable labeled IS.
- 12.
Analytical assay is now ready to test biochemical samples.
- 13.
Run minimum and maximum experiments with calibration curves to determine if the product concentration is in the previously established linear range.
- 14.
Run standard biochemical assay development experiments.
References
- 1.
Slavicek J.M., Hayes-Plazolles N. The Lymantria dispar nucleopolyhedrovirus contains the capsid-associated p24 protein gene.
Virus Genes. 2003;26(1):15–8. [
PubMed: 12680688]
- 2.
Highkin M.K., et al. High-throughput screening assay for sphingosine kinase inhibitors in whole blood using RapidFire(R) mass spectrometry.
J Biomol Screen. 2011;16(2):272–7. [
PubMed: 21297110]
- 3.
Lunn C.A. Label-free screening assays: a strategy for finding better drug candidates.
Future Med Chem. 2010;2(11):1703–16. [
PubMed: 21428840]
- 4.
Jonas M., LaMarr W.A., Ozbal C. Mass spectrometry in high-throughput screening: a case study on acetyl-coenzyme a carboxylase using RapidFire--mass spectrometry (RF-MS).
Comb Chem High Throughput Screen. 2009;12(8):752–9. [
PubMed: 19531010]
- 5.
Hopfgartner G.r., Bourgogne E. Quantitative high-throughput analysis of drugs in biological matrices by mass spectrometry.
Mass Spectrometry Reviews. 2003;22(3):195–214. [
PubMed: 12838545]
- 6.
Beher D., et al. Resveratrol is Not a Direct Activator of SIRT1 Enzyme Activity.
Chemical Biology & Drug Design. 2009;74(6):619–624. [
PubMed: 19843076]
- 7.
- 8.
Morand K.L., et al. Techniques for increasing the throughput of flow injection mass spectrometry.
Anal Chem. 2001;73(2):247–52. [
PubMed: 11199973]
- 9.
Xu R., et al. Application of parallel liquid chromatography/mass spectrometry for high throughput microsomal stability screening of compound libraries.
J Am Soc Mass Spectrom. 2002;13(2):155–65. [
PubMed: 11841071]
- 10.
Quercia A.K., et al. High-throughput screening by mass spectrometry: comparison with the scintillation proximity assay with a focused-file screen of AKT1/PKB alpha.
J Biomol Screen. 2007;12(4):473–80. [
PubMed: 17478485]
- 11.
Annesley T.M. Ion suppression in mass spectrometry.
Clin Chem. 2003;49(7):1041–4. [
PubMed: 12816898]
- 12.
Zhang N.R., et al. Quantitation of small molecules using high-resolution accurate mass spectrometers - a different approach for analysis of biological samples.
Rapid Commun Mass Spectrom. 2009;23(7):1085–94. [
PubMed: 19263405]
- 13.
Masoodi M., Nicolaou A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry.
Rapid Communications in Mass Spectrometry. 2006;20(20):3023–3029. [
PMC free article: PMC1805459] [
PubMed: 16986207]
- 14.
Masoodi M., et al. Simultaneous lipidomic analysis of three families of bioactive lipid mediators leukotrienes, resolvins, protectins and related hydroxy-fatty acids by liquid chromatography/electrospray ionisation tandem mass spectrometry.
Rapid Communications in Mass Spectrometry. 2008;22(2):75–83. [
PMC free article: PMC2542421] [
PubMed: 18059001]
- 15.
Banerjee S., Mazumdar S. Electrospray ionization mass spectrometry: a technique to access the information beyond the molecular weight of the analyte.
Int J Anal Chem. 2012;2012:282574. [
PMC free article: PMC3348530] [
PubMed: 22611397]
- 16.
Liuni P., Wilson D.J. Understanding and optimizing electrospray ionization techniques for proteomic analysis.
Expert Rev Proteomics. 2011;8(2):197–209. [
PubMed: 21501013]
- 17.
Cole, R.B. and Editor, Electrospray And MALDI Mass Spectrometry: Fundamentals, Instrumentation, Practicalities, And Biological Applications, Second Edition. 2010: John Wiley & Sons, Inc. 847 pp.
- 18.
Cooper, B.T., Applied Electrospray Mass Spectrometry. Practical Spectroscopy Series. Volume 32. Edited by Birendra N. Pramanik (Schering-Plough Research Institute, Kenilworth, NJ), A. K. Ganguly (Stevens Institute of Technology, Hoboken, NJ), and Michael L. Gross (Washington University, Saint Louis). Marcel Dekker: New York and Basel. 2002. viii + 434 pp. $185.00. ISBN 0-8247-0618-8. J. Am. Chem. Soc., 2002. 124(Copyright (C) 2012 American Chemical Society (ACS). All Rights Reserved.): p. 13638-13639.
- 19.
West C., Elfakir C., Lafosse M. Porous graphitic carbon: a versatile stationary phase for liquid chromatography.
J Chromatogr A. 2010;1217(19):3201–16. [
PubMed: 19811787]
- 20.
Gerber F., et al. Practical aspects of fast reversed-phase high-performance liquid chromatography using 3 microm particle packed columns and monolithic columns in pharmaceutical development and production working under current good manufacturing practice.
J Chromatogr A. 2004;1036(2):127–33. [
PubMed: 15146913]
- 21.
Roses M., Subirats X., Bosch E. Retention models for ionizable compounds in reversed-phase liquid chromatography: effect of variation of mobile phase composition and temperature.
J Chromatogr A. 2009;1216(10):1756–75. [
PubMed: 19167714]
- 22.
Novakova L., Vlckova H. A review of current trends and advances in modern bio-analytical methods: chromatography and sample preparation.
Anal Chim Acta. 2009;656(1-2):8–35. [
PubMed: 19932811]
- 23.
Lloyd R. Snyder, J.J.K., John W. Dolan, ed. Introduction to Modern Liquid Chromatography 3 rd ed. 2010, John Wiley & Sons, Inc.
- 24.
Lough, W.J., I.W. Wainer, and Editors, High Performance Liquid Chromatography: Fundamental Principles and Practice. 1996: Blackie. 276 pp.
- 25.
Snyder, L.R., et al., Introduction to Modern Liquid Chromatography, Third Edition. 2010: John Wiley & Sons, Inc. 912 pp.
- 26.
Rohrs, H.W., LC/MS: A practical user's guide, by Marvin C. McMaster, Wiley-Interscience Hoboken, NJ, 07030, USA. J. Am. Soc. Mass Spectrom., 2006. 17(Copyright (C) 2012 American Chemical Society (ACS). All Rights Reserved.): p. 1193.
- 27.
- 28.
Hsieh Y. Hydrophilic interaction liquid chromatography-tandem mass spectrometry for drug development.
Curr Drug Discov Technol. 2010;7(3):223–31. [
PubMed: 20843291]
- 29.
Hutchinson S.E., et al. Enabling lead discovery for histone lysine demethylases by high-throughput RapidFire mass spectrometry.
J Biomol Screen. 2012;17(1):39–48. [
PubMed: 21859681]
- 30.
Wu X., et al. In vitro ADME profiling using high-throughput rapidfire mass spectrometry: cytochrome p450 inhibition and metabolic stability assays.
J Biomol Screen. 2012;17(6):761–72. [
PubMed: 22460176]
- 31.
Leveridge M.V., et al. Lead discovery for microsomal prostaglandin E synthase using a combination of high-throughput fluorescent-based assays and RapidFire mass spectrometry.
J Biomol Screen. 2012;17(5):641–50. [
PubMed: 22337655]
- 32.
Briem S., Pettersson B., Skoglund E. Description and validation of a four-channel staggered LC-MS/MS system for high-throughput in vitro screens.
Anal Chem. 2005;77(6):1905–10. [
PubMed: 15762603]
- 33.
Roddy T.P., et al. Mass spectrometric techniques for label-free high-throughput screening in drug discovery.
Anal Chem. 2007;79(21):8207–13. [
PubMed: 17902631]
- 34.
Li, S., et al.,
Full utilization of a mass spectrometer using on-demand sharing with multiple LC units. J. Mass Spectrom., 2012.
47(Copyright (C) 2012 American Chemical Society (ACS). All Rights Reserved.): p. 1074-1082. [
PubMed: 22899517]